- Phylogenetic disorders of respiratory system

Содержание
- 2. PHYLOGENETIC DISORDERS OF RESPIRATORY SYSTEM REPRESENTED BY : DHRUV MANGAL 195 b (LA-2) SUPERVISOR: ANNA ZHUKOVA
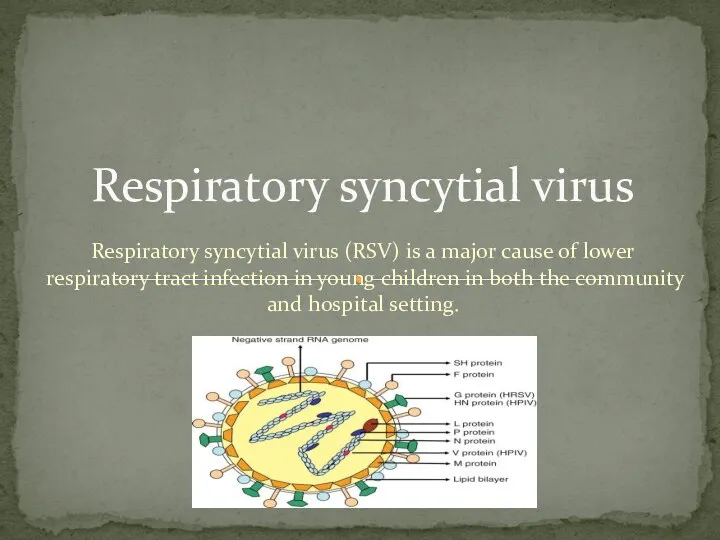
- 3. Respiratory syncytial virus (RSV) is a major cause of lower respiratory tract infection in young children
- 4. Respiratory syncytial virus (RSV) is an important cause of bronchiolitis and pneumonia in infants and young
- 5. A commercial multiplex PCR assay (Seeplex RV7, Seegene, Seoul, South Korea) was used to screen for
- 6. Two-hundred and twenty-six children with PCR-confirmed RSV acute lower respiratory tract infection were identified during the
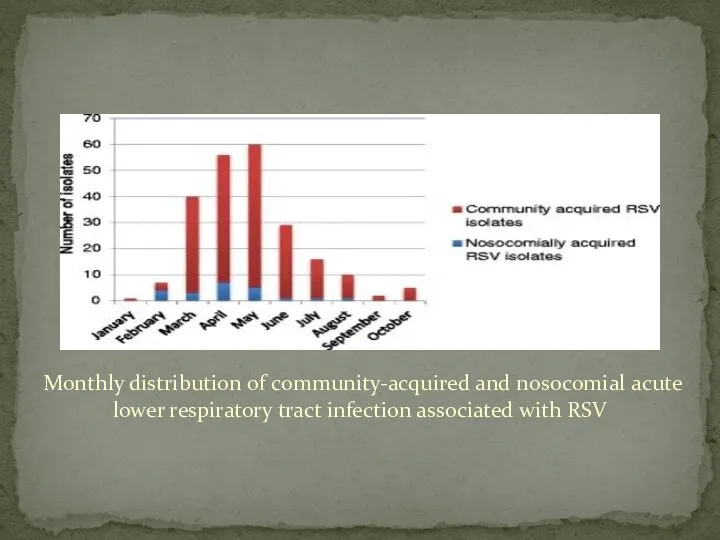
- 7. The median duration of symptoms preceding hospitalisation was 2 days (IQR: 1–4 days). As shown in
- 8. RSV A and RSV B accounted for 181 (80.1 %) and 45 (19.9 %) of the
- 9. Monthly distribution of community-acquired and nosocomial acute lower respiratory tract infection associated with RSV
- 10. Factors significantly associated with nosocomial infection on univariate analysis included age 6 months or older and
- 11. In order to evaluate the circulation of the different human rhinovirus (HRV) species and genotypes in
- 12. The study was carried out in Pediatric Clinic 1 of the Department of Pathophysiology and Transplantation
- 13. Viral nucleic acids were extracted from the nasopharyngeal swabs using a Nuclisens EasyMAG automated extraction system
- 14. The hypervariable part of the 5' NCR (non-coding region), the entire VP4 gene and the 5'
- 15. The HRV sequences showed marked genetic diversity. The HRV-C sequences were the most heterogenous, with an
- 16. Pasteurella multocida is a leading cause of respiratory diseases in many host species. To understand the
- 17. A total of 47 P. multocida strains were selected for whole genome sequencing in this study
- 18. he phylogenetic relationship between P. multocida strains from different host species was predicted by analyzing the
- 20. Whole genome sequencing yielded approximately 796.25~1823.87 Mbp raw data for the 47 porcine P. multocida isolates.
- 22. Скачать презентацию
PHYLOGENETIC DISORDERS OF RESPIRATORY SYSTEM
REPRESENTED BY :
DHRUV MANGAL
195 b (LA-2)
SUPERVISOR:
ANNA
PHYLOGENETIC DISORDERS OF RESPIRATORY SYSTEM
REPRESENTED BY :
DHRUV MANGAL
195 b (LA-2)
SUPERVISOR:
ANNA
Respiratory syncytial virus (RSV) is a major cause of lower respiratory
Respiratory syncytial virus (RSV) is a major cause of lower respiratory

Respiratory syncytial virus (RSV) is an important cause of bronchiolitis and
Respiratory syncytial virus (RSV) is an important cause of bronchiolitis and

A commercial multiplex PCR assay (Seeplex RV7, Seegene, Seoul, South Korea)
A commercial multiplex PCR assay (Seeplex RV7, Seegene, Seoul, South Korea)
Two-hundred and twenty-six children with PCR-confirmed RSV acute lower respiratory tract
Two-hundred and twenty-six children with PCR-confirmed RSV acute lower respiratory tract

The median duration of symptoms preceding hospitalisation was 2 days (IQR:
The median duration of symptoms preceding hospitalisation was 2 days (IQR:

RSV A and RSV B accounted for 181 (80.1 %) and
RSV A and RSV B accounted for 181 (80.1 %) and
Monthly distribution of community-acquired and nosocomial acute lower respiratory tract infection
Monthly distribution of community-acquired and nosocomial acute lower respiratory tract infection

Factors significantly associated with nosocomial infection on univariate analysis included age
Factors significantly associated with nosocomial infection on univariate analysis included age

In order to evaluate the circulation of the different human rhinovirus
In order to evaluate the circulation of the different human rhinovirus

The study was carried out in Pediatric Clinic 1 of the
The study was carried out in Pediatric Clinic 1 of the

Viral nucleic acids were extracted from the nasopharyngeal swabs using a
Viral nucleic acids were extracted from the nasopharyngeal swabs using a

The hypervariable part of the 5' NCR (non-coding region), the entire
The hypervariable part of the 5' NCR (non-coding region), the entire

The HRV sequences showed marked genetic diversity. The HRV-C sequences were
The HRV sequences showed marked genetic diversity. The HRV-C sequences were

Pasteurella multocida is a leading cause of respiratory diseases in many
Pasteurella multocida is a leading cause of respiratory diseases in many

A total of 47 P. multocida strains were selected for whole
A total of 47 P. multocida strains were selected for whole

he phylogenetic relationship between P. multocida strains from different host species
he phylogenetic relationship between P. multocida strains from different host species
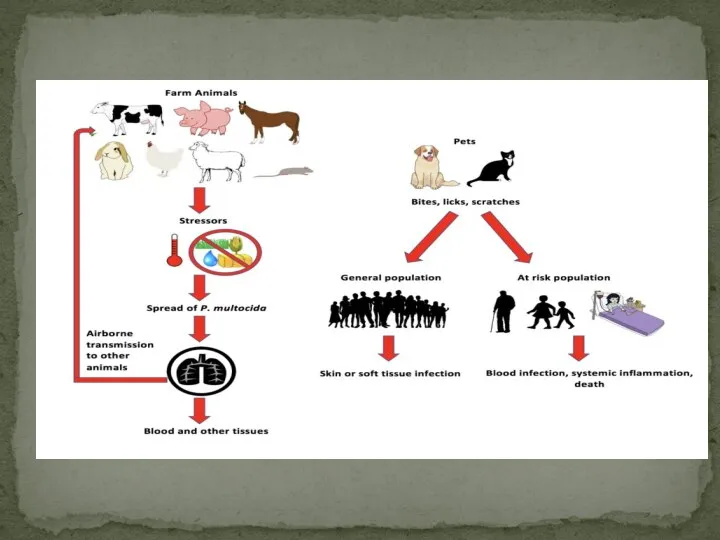

Whole genome sequencing yielded approximately 796.25~1823.87 Mbp raw data for the
Whole genome sequencing yielded approximately 796.25~1823.87 Mbp raw data for the
 Общие правила оказания первой доврачебной помощи. Алгоритм оказания первой помощи. Юридические и моральные аспекты
Общие правила оказания первой доврачебной помощи. Алгоритм оказания первой помощи. Юридические и моральные аспекты Клинические формы вторичного туберкулеза
Клинические формы вторичного туберкулеза Рентгеноконтрастные исследования и препараты
Рентгеноконтрастные исследования и препараты Анатомо-физиологические особенности эндокринной системы у детей
Анатомо-физиологические особенности эндокринной системы у детей Мочекаменная болезнь. Гидронефроз
Мочекаменная болезнь. Гидронефроз Вич и Спид
Вич и Спид Інфузійна терапія
Інфузійна терапія Градация доказательств и уровни рекомендаций
Градация доказательств и уровни рекомендаций Особенности сестринского ухода за инфекционными больными. Сестринский процесс. Сестринский диагноз
Особенности сестринского ухода за инфекционными больными. Сестринский процесс. Сестринский диагноз Анализ затрат на лекарственные средства с помощью ABC/VEV методологии
Анализ затрат на лекарственные средства с помощью ABC/VEV методологии Моногибридті будандастыру. Гибридологиялық зерттеу әдісі
Моногибридті будандастыру. Гибридологиялық зерттеу әдісі Опухоли. Онкология
Опухоли. Онкология Анонимные Наркоманы г. Йошкар-Ола
Анонимные Наркоманы г. Йошкар-Ола Энтеробиоз: определение
Энтеробиоз: определение Ожирение. Степени ожирения
Ожирение. Степени ожирения Генетика человека. Генные болезни
Генетика человека. Генные болезни Острые воспалительные заболевания матки и придатков как причина развития клиники острого живота в гинекологии
Острые воспалительные заболевания матки и придатков как причина развития клиники острого живота в гинекологии Лекарственная токсикология
Лекарственная токсикология Орталық және шеткі жүйке жүйесінің клиникалық физиологиясы бен биохимиясы
Орталық және шеткі жүйке жүйесінің клиникалық физиологиясы бен биохимиясы История развития психогенетики в мировой науке
История развития психогенетики в мировой науке Ас қорыту жүйесіне жалпы шолу
Ас қорыту жүйесіне жалпы шолу Микробиологическая диагностика брюшного тифа, паратифов и других сальмонеллезных инфекций. Пищевые отравления и их диагностика
Микробиологическая диагностика брюшного тифа, паратифов и других сальмонеллезных инфекций. Пищевые отравления и их диагностика Лабораторная диагностика заболеваний, вызываемых извитыми формами бактерий. Спирохетозы (сифилис, лептоспироз, возвратные тифы)
Лабораторная диагностика заболеваний, вызываемых извитыми формами бактерий. Спирохетозы (сифилис, лептоспироз, возвратные тифы) Жарақаттар
Жарақаттар Диагностическая информативность онкомаркеров в гинекологии
Диагностическая информативность онкомаркеров в гинекологии Специфическая (антидотная) фармакотерапия острых отравлений
Специфическая (антидотная) фармакотерапия острых отравлений Эректильная дисфункция (ЭД)
Эректильная дисфункция (ЭД) Лекарственные препараты по химии
Лекарственные препараты по химии