- Прогрессирующие мышечные дистрофии

Содержание
- 2. гетерогенная группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями Прогрессирующие мышечные
- 3. Сарколеммные миопатии: Дистрофинопатии Саркогликанопатии Дистрогликанопатии Кавеолинопатии Дисферлинопатии Плектинопатия Матриксные миопатии: Мерозин-дефицитная Эмеринопатии Ламинопатии Структурные миопатии: Немалиновые
- 4. 1. X-сцепленные мышечные дистрофии 1.1. Миодистрофия Дюшенна и Беккера (дистрофинопатии) 1.1.1. Дистрофинопатия у девочек с синдромом
- 5. 2. Аутосомные мышечные дистрофии 2.1. Лицелопаточно-плечевая Ландузи - Дежерина 2.1.1. Инфантильная форма лицелопаточно-плечевой миодистрофии 2.2. Скапулоперонеальная
- 6. 2.5. Дистальные миодистрофии 2.5.1. Дистальная МД с началом в грудном возрасте 2.5.2. Дистальная МД с началом
- 7. Симметричная проксимальная мышечная слабость, постепенное развитие атрофий Затруднение при ходьбе по лестнице Приемы Говерса при вставании
- 8. симптом свободных надплечий симптом крыловидных лопаток поясничный гиперлордоз, псевдогипертрофии икроножных мышц
- 9. Стадии течения миодистрофического процесса (Бадалян Л. О., 1974) I стадия — с умеренно выраженными двигательными нарушениями
- 10. 1) неблагоприятный вариант — обездвиженность через 5—10 лет от начала болезни, быстрое нарастание нарушения жизнедеятельности, инвалидизации;
- 11. Французский невролог Жульем Бенджамин Аманд Дюшенн впервые описал заболевание в 1861 г. Миодистрофия Дюшенна Guillaume Benjamin
- 12. Наиболее распространенная форма ПМД. Заболеваемость - 13–33 случая на 100 000 родившихся. Тип наследования: АР, Х-
- 13. Схематическое расположение экзонов в гене дистрофина
- 14. Схема делеции 48, 49 и 50-го экзонов в гене дистрофина
- 15. Схема делеции 48, 49, 50 и 51-го экзонов в гене дистрофина
- 16. Дистрофин (более 2 млн нуклеотидов) локализуется на цитоплазматической поверхности сарколеммы мышечного волокна, является частью цитоскелета, обеспечивает
- 17. При сокращении мышечного волокна происходит «скольжение» сократительных белков относительно друг друга, которые должны быть фиксированы к
- 18. Иммунопатологические механизмы: хронический воспалительный процесс и нарушение процессов регенерации. Реакции воспалительного каскада запускаются вскоре в 8-10
- 19. КЛИНИКА ПМД ДЮШЕННА
- 20. Течение ПМД Дюшенна Возраст больного 0 5 10 15 20 25 30 инвалидное кресло - деформации
- 21. Проявляется в возрасте 2—5 лет. Течение быстро прогрессирующее, злокачественное. Обездвиженность наступает в возрасте 14—15 лет, смерть
- 22. Ретракция ахилловых сухожилий не позволяет полноценно опираться на пятки, что определяет ходьбу на пальцах («генеральская походка»)
- 23. Глубокие рефлексы изменяются с различной последовательностью. На ранних стадиях исчезают коленные рефлексы, позже - рефлексы с
- 24. Псевдогипертрофия икроножных мышц создает обманчивое впечатление о сохранности мышечной силы, радует родителей. Псевдогипертрофии мышц могут развиваться
- 25. Атрофии мышц всегда симметричны. Локализуются изначально в проксимальных группах мышц нижних конечностей - мышцах тазового пояса,
- 26. Одна из отличительных особенностей ПМД Дюшенна - сочетание с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой
- 27. Сердечно-сосудистые расстройства (у 73% пациентов) клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением
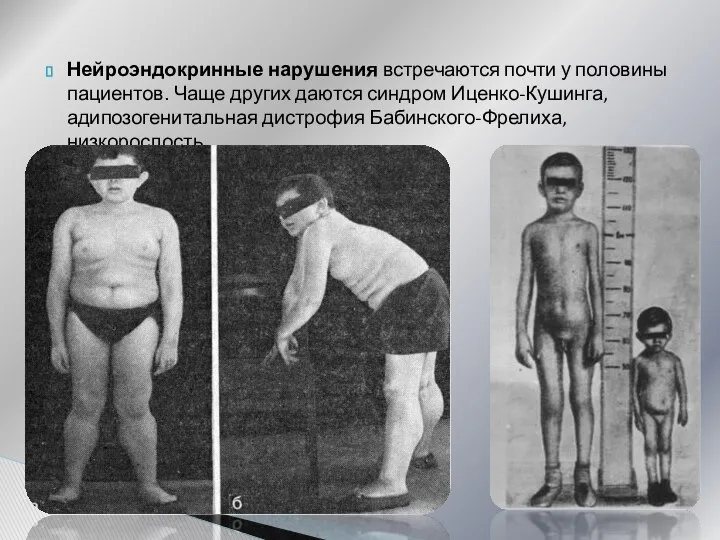
- 28. Нейроэндокринные нарушения встречаются почти у половины пациентов. Чаще других даются синдром Иценко-Кушинга, адипозогенитальная дистрофия Бабинского-Фрелиха, низкорослость.
- 29. Изменения внешности. При слабости и атрофии мышц лица отмечается отсутствие морщин на лбу (симптом «полированного лба»),
- 30. В связи с дефицитом церебральных изоформ дистрофина — аподистрофинов, у 30 % больных с миодистрофией Дюшенна
- 31. Клинические варианты
- 32. Клинические варианты
- 33. Тип наследования: Х-сцепленный, АР дебют от 5 до 20 лет чаще 10-15 лет течение медленно прогрессирующее
- 34. Поясно-конечностная юношеская миодистрофия Эрба — Рота
- 35. АР – тип наследования дебют в детском или юношеском возрасте чаще в 14-16 лет конечностно-поясная миодистрофия
- 36. Формы: ранняя детская, детская и юношеская Характерно поражение гладкой мускулатуры кишечника, развитие сердечно-лёгочной недостаточности, контрактур крупных
- 37. АР – тип наследования дебют чаще к 20 годам, иногда несколько позже слабость и гипотрофия мышц
- 38. Формы ПМД Ландузи-Дежерина: лице-лопаточно-плечевая, лице-лопаточно-плече-перонеальная, лице-лопаточно-плече-ягодично-бедренная, лице-лопаточно-плече-ягодично-бедренно-перонеальная лице-лопаточно-плече-перонеально-ягодично-бедренная. Вариант заболевания с поздним началом (27—30 лет), при
- 39. 1. АД-форма проявляется чаще в детстве, иногда во 2-3-м десятилетии слабость и прогрессирующая гипотрофия мышц плечевого
- 40. Раннее развитие контрактур, чаще в локтевых, коленных суставах, задних мышцах шеи (голова слегка запрокинута) Плечелопаточно-перонеальное распределение
- 41. Основной симптом— хроническая прогрессирующая наружная офтальмоплегия, с присоединением умеренного бульбарного синдрома. В дальнейшем - проксимальная мышечная
- 42. Дистальные миодистрофии Преобладание слабости дистальных отделов При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, При
- 43. Центром по контролю и профилактике заболеваний (Centers for Disease Control and Prevention (CDC)) были разработаны общие
- 44. 1. КФК-MM 2. и-ЭМГ ФВД, ЖЁЛ ЭКГ, ЭХО-КГ ДНК-диагностика Биопсия мышц Диагностика миодистрофии Дюшенна
- 45. Уже в ранних стадиях заболевания обнаруживают креатинурию, гипераминоацидурию, повышение альдолаз, трансаминаз (особенно аланиновой) и специфического фермента
- 46. Повышение уровня КФК (креатинфосфокиназы) отмечается при быстропрогрессирующих формах до 10 000 и выше ммоль/л. Значительное повышение
- 47. На игольчатой ЭМГ – признаки первично-мышечного поражения (потенциаллы фибрилляций и положительные острые волны, выраженность коррелирует с
- 49. В биоптатах — наличие некротизированных мышечных волокон с регенерацией, фагоцитозом и жировым перерождением мышечной ткани. Срез
- 50. Большую информативность имеет УЗИ мышц. Определяется однородность мышечной ткани с равномерным уплотнением и значительным увеличением уровня
- 51. Оценка полиморфизма длины рестрикционных фрагментов – общепринятый стандарт для диагностики болезней Дюшенна выявления носительства гена и
- 52. Исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях, амниоцентез можно использовать после 15
- 53. Вестерн-блоттинг – современный высокочувствительный аналитический метод, используемый для определения специфичных белков в сложных смесях с помощью
- 54. Приём глюкокортикостероидов. Своевременная ГКС-терапия на ранних стадиях задерживает прогрессирование мышечных атрофий, увеличивает мышечную силу и функциое
- 55. В некоторых странах (Великобритания, Испания, Индия, Бразилия, Панама и Гондурас) используется синтетическое прозводное преднизолона – дефлазакорт.
- 56. Приём агонистов β2-адренорецепторов. В нескольких рандомизированных исследованиях показан положительный эффект β2-агонистов (сальбутамол, формотерол и др.) на
- 57. Генотерапевтические подходы: коррекция дефекта путем введения нормальных копий комплементарной ДНК (кДНК) гена дистрофина в составе рекомбинантных
- 58. Трансфекция мышечных волокон с использованием вирусных векторов В экспериментах на мышах удалось продемонстрировать эффективную и трансфекцию
- 59. Невирусные способы доставки кДНК гена дистрофина Невирусные способы доставки включают баллистическую трансфекцию, методы электропорации (электрошока), введение
- 60. Генная терапия на уровне первичного транскрипта гена дистрофина Из этих методов особое внимание привлекает техника направленной
- 61. Активизация экспрессии утрофина – аутосомного гомолога гена дистрофина Суть метода заключается в попытке дерепрессии аутосомного гомолога
- 62. Аминогликозиды. Аминогликозидные антибиотики (в частности, гентамицин) показали свою эффективность в угнетении стоп-кодонов (с появлением которых связано
- 63. Пересадка стволовых клеток. Активно изучаемое направление, пока остающееся в рамках клинического эксперимента. Пересадка миобластов. Её эффективность
- 64. Немедикаментозные методы: Поощрение посильной физической активности (малоподвижный образ жизни ускоряет прогрессирование мышечной дисфункции), дозированная ЛФК с
- 65. При доброкачественных формах миодистрофий в стадии компенсации возможно проведение оперативного вмешательства, направленного на предупреждение и избавление
- 66. Если против какой-нибудь болезни предлагается очень много средств, значит, болезнь неизлечима. А.П.Чехов
- 67. Для Силы, Независимости и Жизни
- 69. Скачать презентацию

гетерогенная группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией
гетерогенная группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией
Сарколеммные миопатии:
Дистрофинопатии
Саркогликанопатии
Дистрогликанопатии
Кавеолинопатии
Дисферлинопатии
Плектинопатия
Матриксные миопатии:
Мерозин-дефицитная
Эмеринопатии
Ламинопатии
Структурные миопатии:
Немалиновые миопатии
Титиновые миопатии
Миотилиновые миопатии
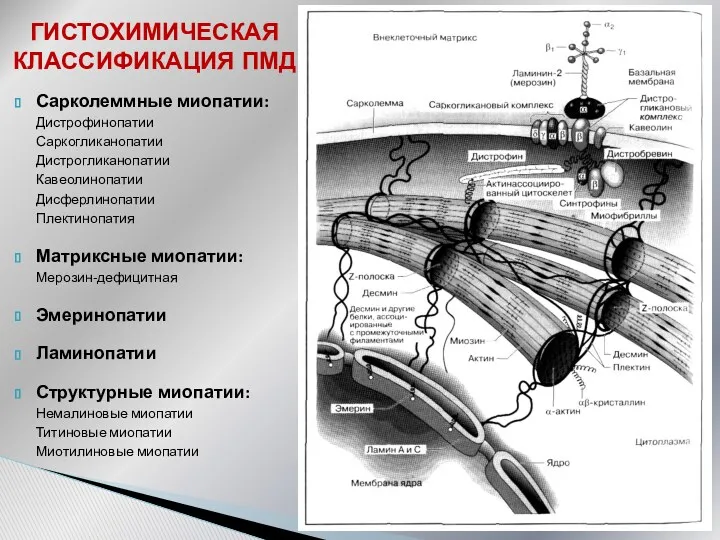
ГИСТОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ ПМД
Сарколеммные миопатии:
Дистрофинопатии
Саркогликанопатии
Дистрогликанопатии
Кавеолинопатии
Дисферлинопатии
Плектинопатия
Матриксные миопатии:
Мерозин-дефицитная
Эмеринопатии
Ламинопатии
Структурные миопатии:
Немалиновые миопатии
Титиновые миопатии
Миотилиновые миопатии
ГИСТОХИМИЧЕСКАЯ КЛАССИФИКАЦИЯ ПМД

1. X-сцепленные мышечные дистрофии
1.1. Миодистрофия Дюшенна и Беккера (дистрофинопатии)
1.1.1. Дистрофинопатия у
1.1. Миодистрофия Дюшенна и Беккера (дистрофинопатии)
1.1.1. Дистрофинопатия у

2. Аутосомные мышечные дистрофии
2.1. Лицелопаточно-плечевая Ландузи - Дежерина
2.1.1. Инфантильная
2. Аутосомные мышечные дистрофии
2.1. Лицелопаточно-плечевая Ландузи - Дежерина
2.1.1. Инфантильная

2.5. Дистальные миодистрофии
2.5.1. Дистальная МД с началом в грудном возрасте
2.5.2. Дистальная
2.5.1. Дистальная МД с началом в грудном возрасте
2.5.2. Дистальная

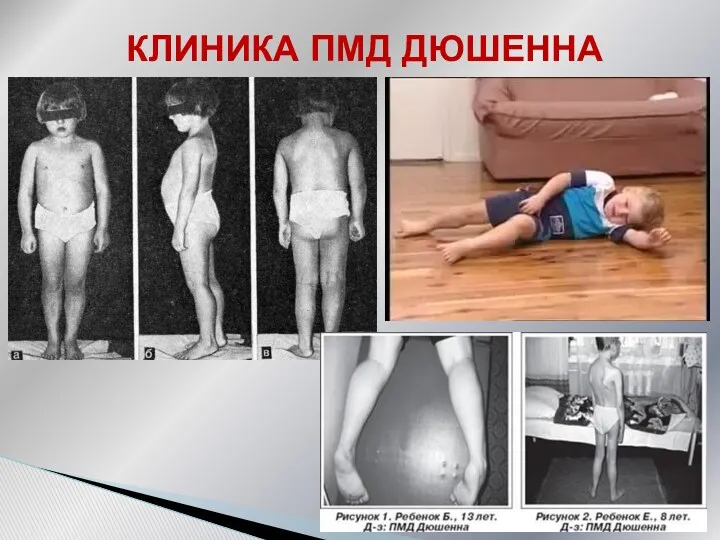
Симметричная проксимальная мышечная слабость, постепенное развитие атрофий
Затруднение при ходьбе по лестнице
Приемы
Симметричная проксимальная мышечная слабость, постепенное развитие атрофий
Затруднение при ходьбе по лестнице
Приемы
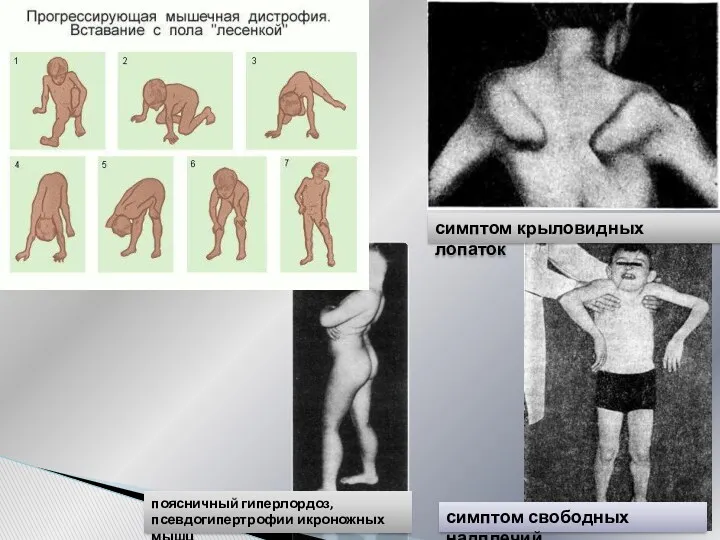
симптом свободных надплечий
симптом крыловидных лопаток
поясничный гиперлордоз,
псевдогипертрофии икроножных мышц
симптом свободных надплечий
симптом крыловидных лопаток
поясничный гиперлордоз,
псевдогипертрофии икроножных мышц

Стадии течения миодистрофического процесса
(Бадалян Л. О., 1974)
I стадия — с
Стадии течения миодистрофического процесса
(Бадалян Л. О., 1974)
I стадия — с

1) неблагоприятный вариант — обездвиженность через 5—10 лет от начала болезни,

Французский невролог Жульем Бенджамин Аманд Дюшенн впервые описал заболевание в 1861
Французский невролог Жульем Бенджамин Аманд Дюшенн впервые описал заболевание в 1861

Наиболее распространенная форма ПМД.
Заболеваемость - 13–33 случая на 100 000
Наиболее распространенная форма ПМД.
Заболеваемость - 13–33 случая на 100 000

Схематическое расположение экзонов в гене дистрофина
Схематическое расположение экзонов в гене дистрофина
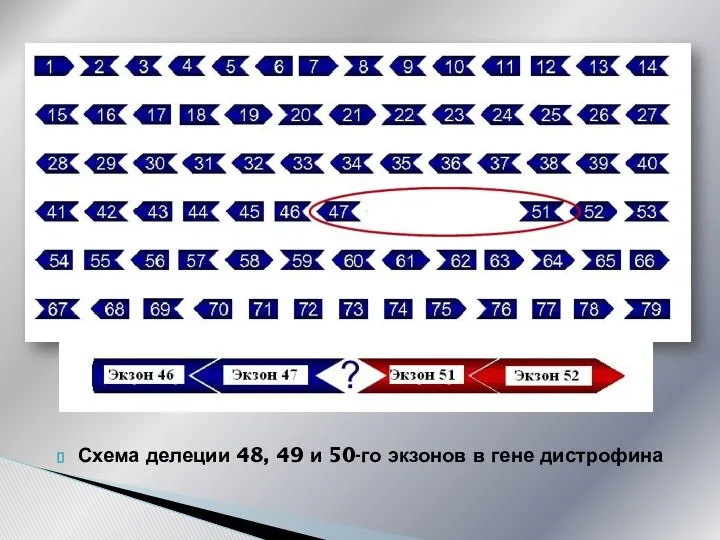
Схема делеции 48, 49 и 50-го экзонов в гене дистрофина
Схема делеции 48, 49 и 50-го экзонов в гене дистрофина

Схема делеции 48, 49, 50 и 51-го экзонов в гене дистрофина
Схема делеции 48, 49, 50 и 51-го экзонов в гене дистрофина
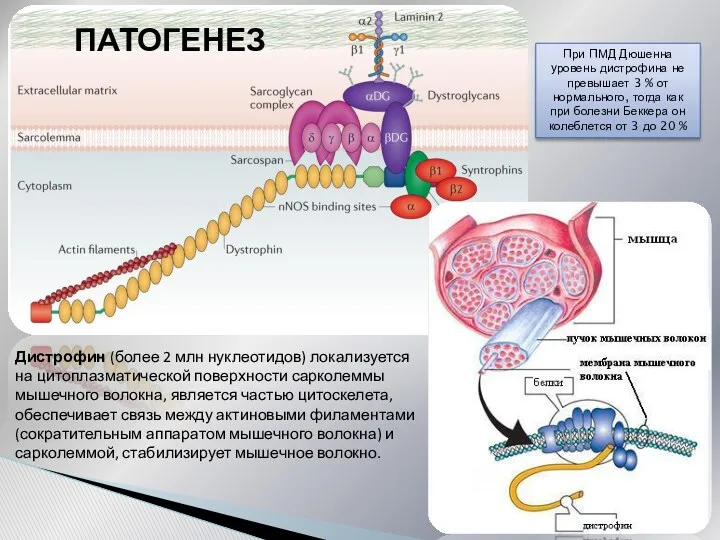
Дистрофин (более 2 млн нуклеотидов) локализуется на цитоплазматической поверхности сарколеммы мышечного
Дистрофин (более 2 млн нуклеотидов) локализуется на цитоплазматической поверхности сарколеммы мышечного

При сокращении мышечного волокна происходит «скольжение» сократительных белков относительно друг друга,

Иммунопатологические механизмы: хронический воспалительный процесс и нарушение процессов регенерации.
Реакции воспалительного
Иммунопатологические механизмы: хронический воспалительный процесс и нарушение процессов регенерации.
Реакции воспалительного
КЛИНИКА ПМД ДЮШЕННА
КЛИНИКА ПМД ДЮШЕННА

Течение ПМД Дюшенна
Возраст больного
0
5
10
15
20
25
30
инвалидное кресло - деформации скелета
трудности при ходьбе
снижение силы
Течение ПМД Дюшенна
Возраст больного
0
5
10
15
20
25
30
инвалидное кресло - деформации скелета
трудности при ходьбе
снижение силы

Проявляется в возрасте 2—5 лет.
Течение быстро прогрессирующее, злокачественное.
Обездвиженность наступает
Проявляется в возрасте 2—5 лет.
Течение быстро прогрессирующее, злокачественное.
Обездвиженность наступает

Ретракция ахилловых сухожилий не позволяет полноценно опираться на пятки, что определяет
Ретракция ахилловых сухожилий не позволяет полноценно опираться на пятки, что определяет

Глубокие рефлексы изменяются с различной последовательностью. На ранних стадиях исчезают коленные
Глубокие рефлексы изменяются с различной последовательностью. На ранних стадиях исчезают коленные
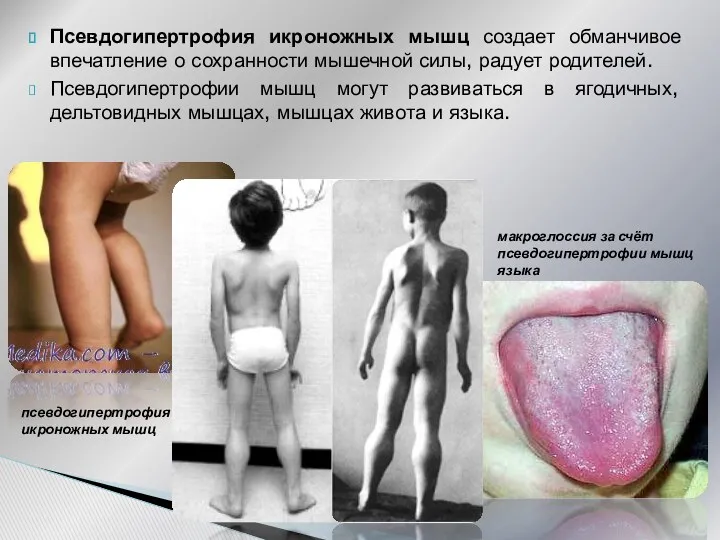
Псевдогипертрофия икроножных мышц создает обманчивое впечатление о сохранности мышечной силы, радует
Псевдогипертрофия икроножных мышц создает обманчивое впечатление о сохранности мышечной силы, радует

Атрофии мышц всегда симметричны.
Локализуются изначально в проксимальных группах мышц нижних
Атрофии мышц всегда симметричны.
Локализуются изначально в проксимальных группах мышц нижних
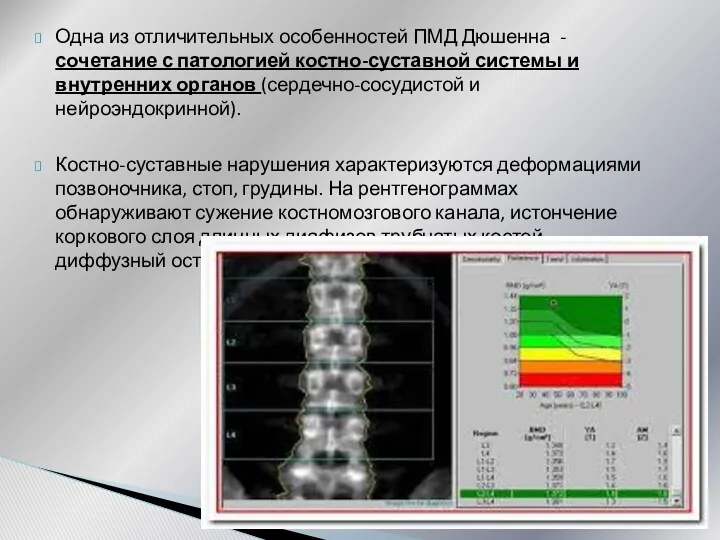
Одна из отличительных особенностей ПМД Дюшенна - сочетание с патологией костно-суставной
Одна из отличительных особенностей ПМД Дюшенна - сочетание с патологией костно-суставной
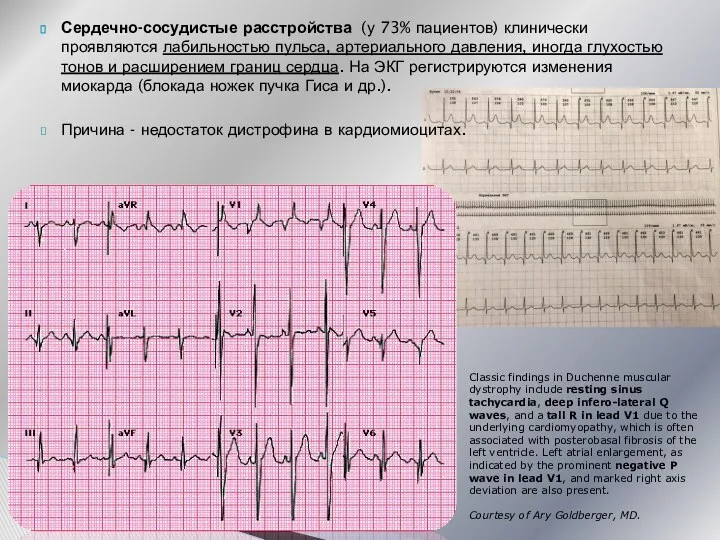
Сердечно-сосудистые расстройства (у 73% пациентов) клинически проявляются лабильностью пульса, артериального давления,
Сердечно-сосудистые расстройства (у 73% пациентов) клинически проявляются лабильностью пульса, артериального давления,
Нейроэндокринные нарушения встречаются почти у половины пациентов. Чаще других даются синдром
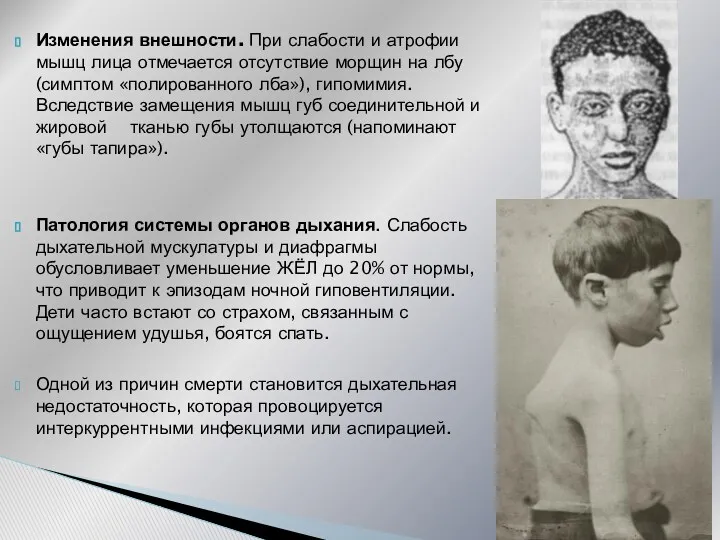
Изменения внешности. При слабости и атрофии мышц лица отмечается отсутствие морщин на
Изменения внешности. При слабости и атрофии мышц лица отмечается отсутствие морщин на

В связи с дефицитом церебральных изоформ дистрофина — аподистрофинов, у 30
В связи с дефицитом церебральных изоформ дистрофина — аподистрофинов, у 30
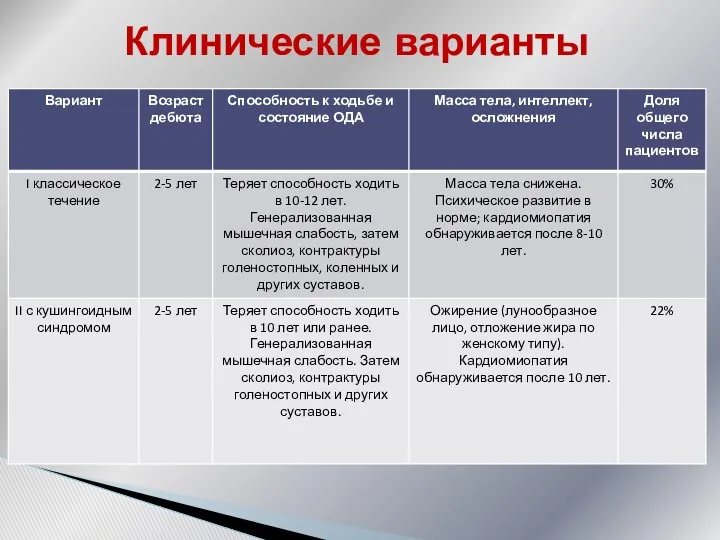
Клинические варианты
Клинические варианты

Клинические варианты
Клинические варианты

Тип наследования: Х-сцепленный, АР
дебют от 5 до 20 лет чаще
Тип наследования: Х-сцепленный, АР
дебют от 5 до 20 лет чаще
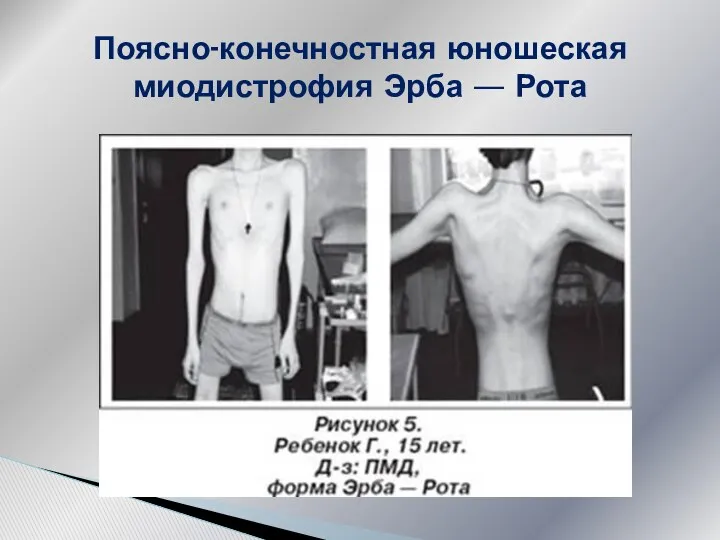
Поясно-конечностная юношеская миодистрофия Эрба — Рота
Поясно-конечностная юношеская миодистрофия Эрба — Рота

АР – тип наследования
дебют в детском или юношеском возрасте чаще в
АР – тип наследования
дебют в детском или юношеском возрасте чаще в

Формы: ранняя детская, детская и юношеская
Характерно поражение гладкой мускулатуры кишечника,
Характерно поражение гладкой мускулатуры кишечника,

АР – тип наследования
дебют чаще к 20 годам, иногда несколько
АР – тип наследования
дебют чаще к 20 годам, иногда несколько
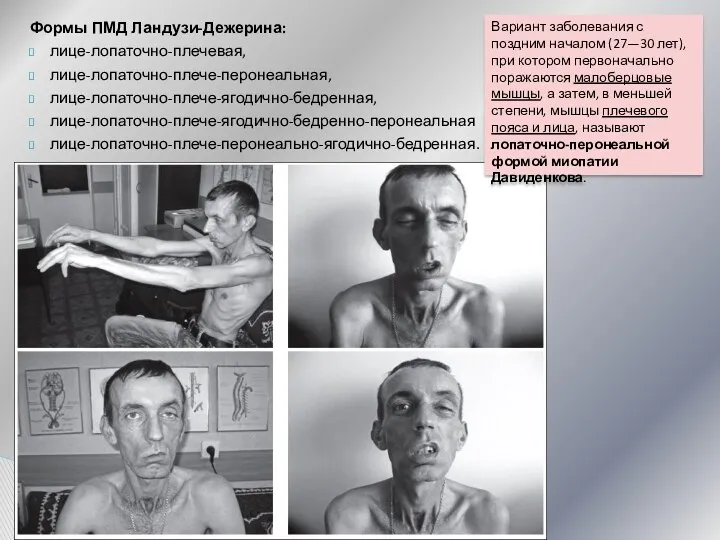
Формы ПМД Ландузи-Дежерина:
лице-лопаточно-плечевая,
лице-лопаточно-плече-перонеальная,
лице-лопаточно-плече-ягодично-бедренная,
лице-лопаточно-плече-ягодично-бедренно-перонеальная
лице-лопаточно-плече-перонеально-ягодично-бедренная.
Вариант заболевания с поздним
Формы ПМД Ландузи-Дежерина:
лице-лопаточно-плечевая,
лице-лопаточно-плече-перонеальная,
лице-лопаточно-плече-ягодично-бедренная,
лице-лопаточно-плече-ягодично-бедренно-перонеальная
лице-лопаточно-плече-перонеально-ягодично-бедренная.
Вариант заболевания с поздним

1. АД-форма
проявляется чаще в детстве, иногда во 2-3-м десятилетии
слабость и прогрессирующая
1. АД-форма
проявляется чаще в детстве, иногда во 2-3-м десятилетии
слабость и прогрессирующая

Раннее развитие контрактур, чаще в локтевых, коленных суставах, задних мышцах шеи
Раннее развитие контрактур, чаще в локтевых, коленных суставах, задних мышцах шеи

Основной симптом— хроническая прогрессирующая наружная офтальмоплегия, с присоединением умеренного бульбарного синдрома.
Основной симптом— хроническая прогрессирующая наружная офтальмоплегия, с присоединением умеренного бульбарного синдрома.
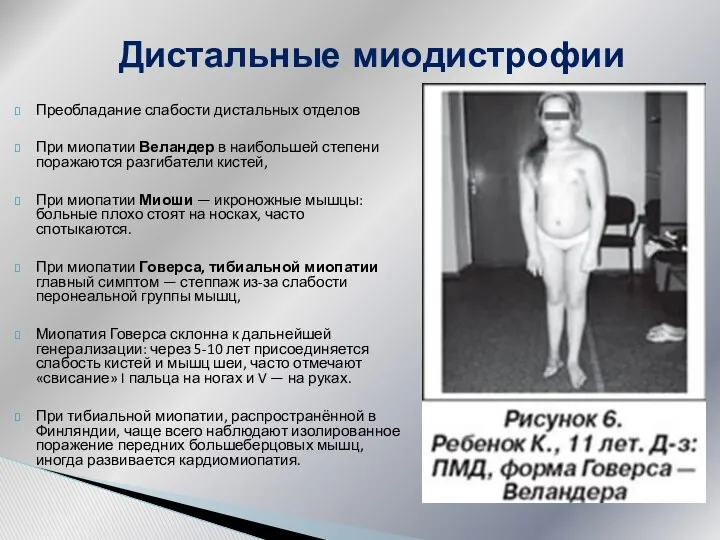
Дистальные миодистрофии
Преобладание слабости дистальных отделов
При миопатии Веландер в наибольшей степени поражаются
Дистальные миодистрофии
Преобладание слабости дистальных отделов
При миопатии Веландер в наибольшей степени поражаются

Центром по контролю и профилактике заболеваний (Centers for Disease Control and
Центром по контролю и профилактике заболеваний (Centers for Disease Control and

1. КФК-MM
2. и-ЭМГ
ФВД, ЖЁЛ
ЭКГ, ЭХО-КГ
ДНК-диагностика
Биопсия мышц
Диагностика миодистрофии Дюшенна
1. КФК-MM
2. и-ЭМГ
ФВД, ЖЁЛ
ЭКГ, ЭХО-КГ
ДНК-диагностика
Биопсия мышц
Диагностика миодистрофии Дюшенна

Уже в ранних стадиях заболевания обнаруживают креатинурию, гипераминоацидурию, повышение альдолаз, трансаминаз
Уже в ранних стадиях заболевания обнаруживают креатинурию, гипераминоацидурию, повышение альдолаз, трансаминаз
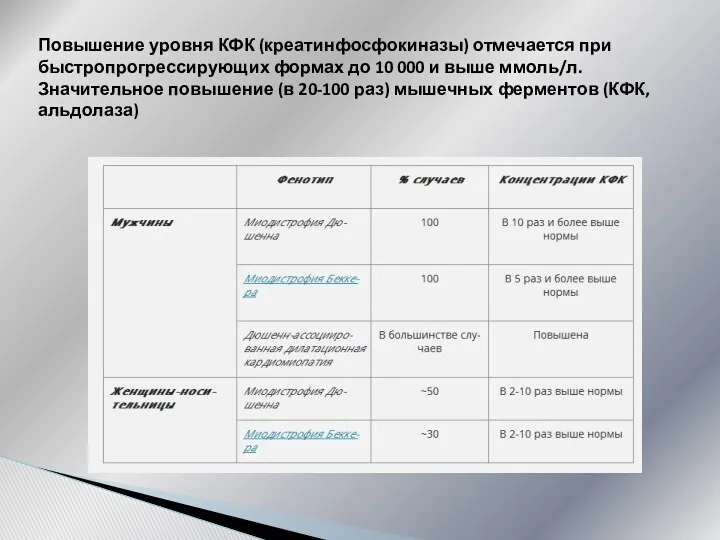
Повышение уровня КФК (креатинфосфокиназы) отмечается при быстропрогрессирующих формах до 10 000
Повышение уровня КФК (креатинфосфокиназы) отмечается при быстропрогрессирующих формах до 10 000

На игольчатой ЭМГ – признаки первично-мышечного поражения (потенциаллы фибрилляций и положительные
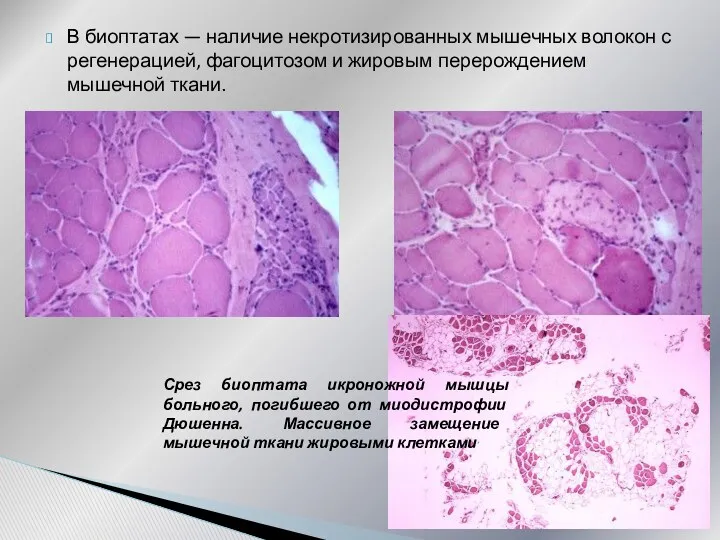
В биоптатах — наличие некротизированных мышечных волокон с регенерацией, фагоцитозом и
В биоптатах — наличие некротизированных мышечных волокон с регенерацией, фагоцитозом и
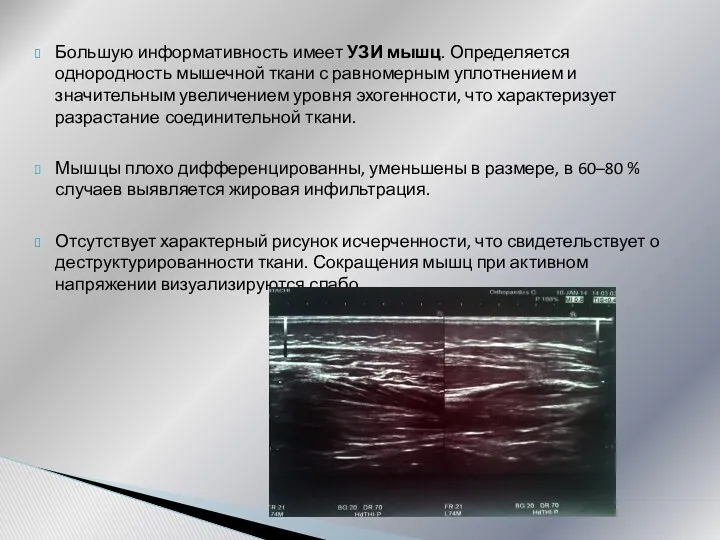
Большую информативность имеет УЗИ мышц. Определяется однородность мышечной ткани с равномерным
Большую информативность имеет УЗИ мышц. Определяется однородность мышечной ткани с равномерным

Оценка полиморфизма длины рестрикционных фрагментов – общепринятый стандарт для диагностики болезней
Оценка полиморфизма длины рестрикционных фрагментов – общепринятый стандарт для диагностики болезней

Исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях,
Исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях,

Вестерн-блоттинг – современный высокочувствительный аналитический метод, используемый для определения специфичных белков в
Вестерн-блоттинг – современный высокочувствительный аналитический метод, используемый для определения специфичных белков в

Приём глюкокортикостероидов. Своевременная ГКС-терапия на ранних стадиях задерживает прогрессирование мышечных атрофий, увеличивает
Приём глюкокортикостероидов. Своевременная ГКС-терапия на ранних стадиях задерживает прогрессирование мышечных атрофий, увеличивает

В некоторых странах (Великобритания, Испания, Индия, Бразилия, Панама и Гондурас) используется
В некоторых странах (Великобритания, Испания, Индия, Бразилия, Панама и Гондурас) используется

Приём агонистов β2-адренорецепторов. В нескольких рандомизированных исследованиях показан положительный эффект β2-агонистов (сальбутамол,
Приём агонистов β2-адренорецепторов. В нескольких рандомизированных исследованиях показан положительный эффект β2-агонистов (сальбутамол,

Генотерапевтические подходы:
коррекция дефекта путем введения нормальных копий комплементарной ДНК (кДНК)
Генотерапевтические подходы:
коррекция дефекта путем введения нормальных копий комплементарной ДНК (кДНК)

Трансфекция мышечных волокон с использованием вирусных векторов
В экспериментах на мышах удалось
Трансфекция мышечных волокон с использованием вирусных векторов
В экспериментах на мышах удалось

Невирусные способы доставки кДНК гена дистрофина
Невирусные способы доставки включают баллистическую трансфекцию,
Невирусные способы доставки кДНК гена дистрофина
Невирусные способы доставки включают баллистическую трансфекцию,

Генная терапия на уровне первичного транскрипта гена дистрофина
Из этих методов особое
Генная терапия на уровне первичного транскрипта гена дистрофина
Из этих методов особое

Активизация экспрессии утрофина – аутосомного гомолога гена дистрофина
Суть метода заключается в
Активизация экспрессии утрофина – аутосомного гомолога гена дистрофина
Суть метода заключается в

Аминогликозиды. Аминогликозидные антибиотики (в частности, гентамицин) показали свою эффективность в угнетении стоп-кодонов
Аминогликозиды. Аминогликозидные антибиотики (в частности, гентамицин) показали свою эффективность в угнетении стоп-кодонов

Пересадка стволовых клеток. Активно изучаемое направление, пока остающееся в рамках клинического эксперимента.
Пересадка стволовых клеток. Активно изучаемое направление, пока остающееся в рамках клинического эксперимента.

Немедикаментозные методы:
Поощрение посильной физической активности (малоподвижный образ жизни ускоряет прогрессирование
Немедикаментозные методы:
Поощрение посильной физической активности (малоподвижный образ жизни ускоряет прогрессирование
При доброкачественных формах миодистрофий в стадии компенсации возможно проведение оперативного вмешательства,
При доброкачественных формах миодистрофий в стадии компенсации возможно проведение оперативного вмешательства,

Если против какой-нибудь болезни предлагается очень много средств, значит, болезнь неизлечима.
Если против какой-нибудь болезни предлагается очень много средств, значит, болезнь неизлечима.
Для Силы, Независимости и Жизни
Для Силы, Независимости и Жизни
 Этиология и патогенез болезней пародонта
Этиология и патогенез болезней пародонта Жүкті әйелдерді амбулаториялық жағдайда жүргізу
Жүкті әйелдерді амбулаториялық жағдайда жүргізу Rak piersi. Leczenie systemowe
Rak piersi. Leczenie systemowe 170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy
170 years since the birth of Ivan Pavlov. Traditions and innovations in Cognitive Behavioral Therapy Оқушының дұрыс тамақтануы - жаңа оқу жылындағы жетістіктердің негізі
Оқушының дұрыс тамақтануы - жаңа оқу жылындағы жетістіктердің негізі Рецепт. Структура рецепта
Рецепт. Структура рецепта Особенности проведения медицинских осмотров обучающихся в целях раннего выявления потребления наркотических средств
Особенности проведения медицинских осмотров обучающихся в целях раннего выявления потребления наркотических средств Ротавирусная инфекция
Ротавирусная инфекция Классификация акушерских кровотечений во время беременности
Классификация акушерских кровотечений во время беременности Оказание первой помощи. Современные требования. Часть 1
Оказание первой помощи. Современные требования. Часть 1 Сүйектің сынуы. Буын шығуы соғып алу. Сіңір созылуы
Сүйектің сынуы. Буын шығуы соғып алу. Сіңір созылуы Гостра серцева недостатність
Гостра серцева недостатність Кандидоз. Причины кандидоза
Кандидоз. Причины кандидоза Старение иммунной системы и ее взаимосвязь с течением коронавирусной инфекции (COVID-19)
Старение иммунной системы и ее взаимосвязь с течением коронавирусной инфекции (COVID-19) Современные лекарственные формы и системы доставки лекарственных средств
Современные лекарственные формы и системы доставки лекарственных средств Современные направления комплексного ухода за пациентами с язвенной болезнью 12-ти перстной кишки
Современные направления комплексного ухода за пациентами с язвенной болезнью 12-ти перстной кишки Острая гнойная инфекция костей и суставов
Острая гнойная инфекция костей и суставов Icon. Современный метод лечения кариеса
Icon. Современный метод лечения кариеса Сестринские манипуляции. Постановка очистительной клизмы
Сестринские манипуляции. Постановка очистительной клизмы Анатомия опорно-двигательного аппарата
Анатомия опорно-двигательного аппарата Тамақтану физиологиясы курсының пәні мен міндеттері
Тамақтану физиологиясы курсының пәні мен міндеттері Задержка роста, не связанная с патологией гипофиза
Задержка роста, не связанная с патологией гипофиза Основные виды патологий зрительного нерва
Основные виды патологий зрительного нерва Иммуностимуляторы
Иммуностимуляторы Скульптор среди медиков. Всё о профессии зубного техника
Скульптор среди медиков. Всё о профессии зубного техника Диспансеризация населения
Диспансеризация населения Вскармливание детей первого года жизни
Вскармливание детей первого года жизни Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2)
Новости nCov-2019 (c 11/02 – COVID-2019, SARS CoV-2)