наследования), клинических обострений болезни (раннее начало в 1—3 года, симметричные атрофии проксимальных групп мышц, развивающиеся в восходящем направлении, псевдогипертрофии икроножных мышц, грубые соматические и нейроэндокринные расстройства, снижение интеллекта, быстрое злокачественное течение болезни), данных биохимических исследований (типично раннее, с 5-го дня жизни ребенка, увеличение активности КФК – в 30—50 раз выше нормы), игольчатой электромиографии и морфологических результатов. позволяющих выявить первично-мышечный (миодистрофический) тип поражения.
Лечение:Радикального лечения миодистрофии Дюшенна пока не предложено.
Существует только симптоматическое лечение. Стероиды способны замедлить развитие болезни. Рекомендована лечебная физкультура. У больных повышен риск переломов костей. Требуется особая осторожность при назначении анестезии (наркоза) при хирургических вмешательствах.
В сентябре 2016 года FDA одобрило первый специализированный препарат — «Эксондис 51» (Exondys 51, этеплирсен), предназначенный для терапии мышечной дистрофии Дюшенна у пациентов с подтвержденной мутацией гена дистрофина, подходящей для корректировки посредством пропуска экзона 51. Таких больных насчитывается приблизительно 13% от общей популяции. Этеплирсен (eteplirsen), разработанный «Сарепта терапьютикс» (Sarepta Therapeutics), представляет собой антисмысловой олигонуклеотид, который связывается с экзоном 51 пре-мРНК дистрофина, чтобы исключать этот экзон в ходе мРНК-процессинга, тем самым реализуя альтернативный сплайсинг с последующим синтезом внутренне усеченного, но функционального дистрофина. Впрочем, терапевтическая эффективность этеплирсена в виде улучшения моторных функций по-прежнему остается недоказанной.

 Классификация психоактивных веществ
Классификация психоактивных веществ Дәрігердің кәсіптік деформациясы
Дәрігердің кәсіптік деформациясы Туберкулезді менингит
Туберкулезді менингит Профили суточной динамики АД и их клиническое значение
Профили суточной динамики АД и их клиническое значение Фемофлор-скрининг. Биоценоз влагалища
Фемофлор-скрининг. Биоценоз влагалища Сахарный диабет. Заболевания щитовидной железы. Ожирение
Сахарный диабет. Заболевания щитовидной железы. Ожирение Потребность пациента в нормальном дыхании и кровообращении
Потребность пациента в нормальном дыхании и кровообращении Общая гигиена
Общая гигиена Психология здоровья личности
Психология здоровья личности Ортанғы құлақтың созылмалы қабынуы. Бассүйекішілік отогенді асқыну
Ортанғы құлақтың созылмалы қабынуы. Бассүйекішілік отогенді асқыну Организация психиатрической помощи в РФ
Организация психиатрической помощи в РФ Поражение почек при первичном и вторичном антифосфолипидном синдроме
Поражение почек при первичном и вторичном антифосфолипидном синдроме Метаболический синдром
Метаболический синдром Бабезіоз великої рогатої худоби
Бабезіоз великої рогатої худоби Старость. Старение организма. (Лекция 37)
Старость. Старение организма. (Лекция 37) Острые кишечные инфекции у детей
Острые кишечные инфекции у детей Иммуномодуляторлар. Иммуностимулдеуші
Иммуномодуляторлар. Иммуностимулдеуші Анемии. (Лекция 2)
Анемии. (Лекция 2) Чума. Симптомы. Клиника. Лечение
Чума. Симптомы. Клиника. Лечение Лечение заболеваний ЖКТ у детей. Стоматиты, гастрит и гастродуоденит, язвенная болезнь, панкреатит
Лечение заболеваний ЖКТ у детей. Стоматиты, гастрит и гастродуоденит, язвенная болезнь, панкреатит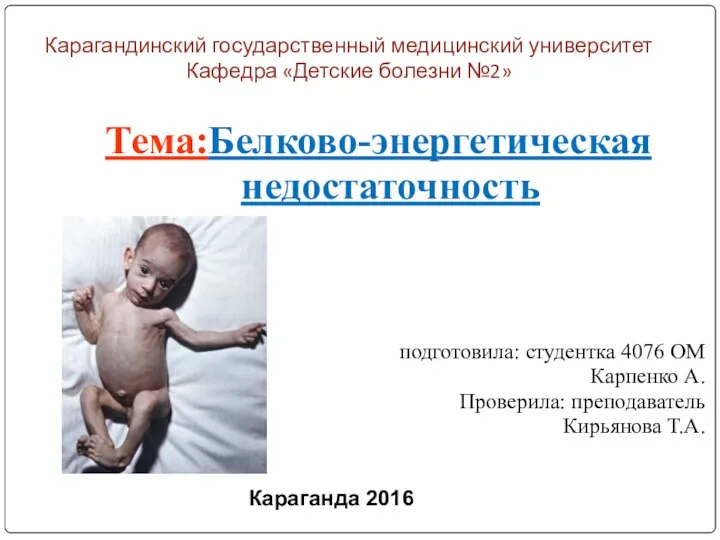 Белково-энергетическая недостаточность
Белково-энергетическая недостаточность Рак гортани. Диагностика. Лечение. Прогноз
Рак гортани. Диагностика. Лечение. Прогноз Буллезный эпидермолиз
Буллезный эпидермолиз Бронхиальді астма диагностикасы және дифференциалды диагностикасы
Бронхиальді астма диагностикасы және дифференциалды диагностикасы Обструктивный бронхит. Дифференциальная диагностика обструктивного синдрома
Обструктивный бронхит. Дифференциальная диагностика обструктивного синдрома Артериальді гипертензия
Артериальді гипертензия Лекарственные растения и сырье, содержащие витамины
Лекарственные растения и сырье, содержащие витамины Эпидемический процесс
Эпидемический процесс