- Синдром MELAS

Содержание
- 2. ЧТО ТАКОЕ MELAS? ME - Mitochondrial Encephalomyopathy ( митохондриальная энцефаломиопатия) LA - Lactic Acidosis (лактатацидоз) S
- 3. ЧТО ТАКОЕ MELAS? Синдром MELAS— прогрессирующее нейродегенеративное заболевание. В каждом конкретном случае набор симптомов и их
- 4. РАСПРОСТРАНЕННОСТЬ Среди взрослого населения Финляндии число лиц с синдромом и мутацией A3243G было оценено в 10.2
- 5. ПРИЧИНЫ ЗАБОЛЕВАНИЯ У большинства пациентов (80%) синдром MELAS обусловлен точковой заменой A3243G в гене тРНК лейцина
- 6. ПАТОГЕНЕЗ Мутации мтДНК, контролирующие дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования - важнейшего источника энергии
- 7. СХЕМА ПОЛУЧЕНИЯ ЭНЕРГИИ КЛЕТКОЙ
- 8. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ Возраст: 5-35 лет (чаще 5-15) Манифестация: инсульто-подобные состояния (кортикальный или субкортикальный инфаркт), злокачественная мигрень.
- 9. Неврологические симптомы : эпилептические приступы ( фокальные, вторично генерализованные, диалептические и другие), мозжечковые расстройства, миоклонус-эпилепсия, корковая
- 10. Нейропатологические изменения головного мозга включают гибель нейронов, демиелинизацию, пролиферацию астроцитов. Кальцификаты в области базальных ганглиев, нередко
- 11. Мышечная слабость, нейросенсорная тугоухость являются типичными симптомами заболевания. Эндокринопатии могут быть представлены недостаточностью гормона роста, сахарным
- 12. Характерен значительный уровень лактат-ацидоза в крови и спинномозговой жидкости, при биопсии скелетных мышц нередко выявляется феномен
- 13. ОСОБЕННОСТИ ТЕЧЕНИЯ Больные низкорослы Мутация не всегда ведет в заболеванию (из-за гетероплазмии большинство митохондрий у человека
- 14. ДИАГНОСТИКА Биохимические, морфологические и молекулярногенетические методы. У всех пациентов с синдромом MELAS выявляют повышение лактата и
- 16. Скачать презентацию

ЧТО ТАКОЕ MELAS?
ME - Mitochondrial Encephalomyopathy ( митохондриальная энцефаломиопатия)
LA -
ЧТО ТАКОЕ MELAS?
ME - Mitochondrial Encephalomyopathy ( митохондриальная энцефаломиопатия)
LA -
ЧТО ТАКОЕ MELAS?
Синдром MELAS— прогрессирующее нейродегенеративное заболевание.
В каждом конкретном случае
ЧТО ТАКОЕ MELAS?
Синдром MELAS— прогрессирующее нейродегенеративное заболевание.
В каждом конкретном случае

РАСПРОСТРАНЕННОСТЬ
Среди взрослого населения Финляндии число лиц с синдромом и мутацией A3243G
РАСПРОСТРАНЕННОСТЬ
Среди взрослого населения Финляндии число лиц с синдромом и мутацией A3243G

ПРИЧИНЫ ЗАБОЛЕВАНИЯ
У большинства пациентов (80%) синдром MELAS обусловлен точковой заменой
ПРИЧИНЫ ЗАБОЛЕВАНИЯ
У большинства пациентов (80%) синдром MELAS обусловлен точковой заменой

ПАТОГЕНЕЗ
Мутации мтДНК, контролирующие дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования
ПАТОГЕНЕЗ
Мутации мтДНК, контролирующие дыхательную цепь митохондрий, сопровождаются нарушением процессов окислительного фосфорилирования
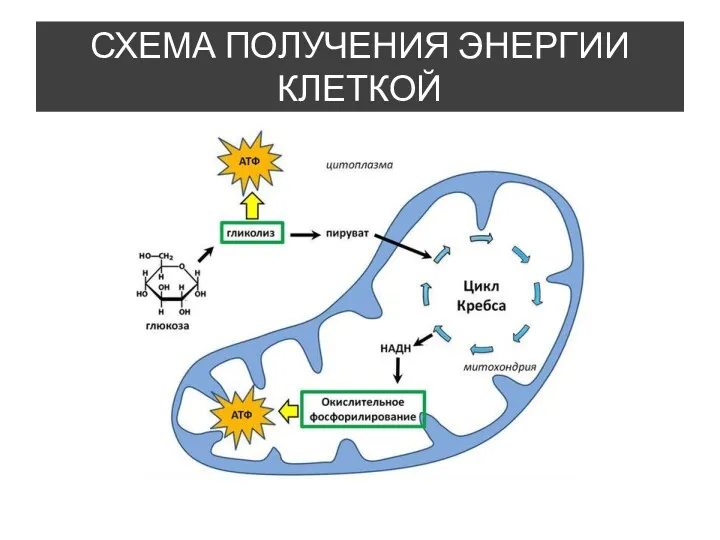
СХЕМА ПОЛУЧЕНИЯ ЭНЕРГИИ КЛЕТКОЙ
СХЕМА ПОЛУЧЕНИЯ ЭНЕРГИИ КЛЕТКОЙ

КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Возраст: 5-35 лет (чаще 5-15)
Манифестация:
инсульто-подобные состояния
(кортикальный
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Возраст: 5-35 лет (чаще 5-15)
Манифестация:
инсульто-подобные состояния
(кортикальный
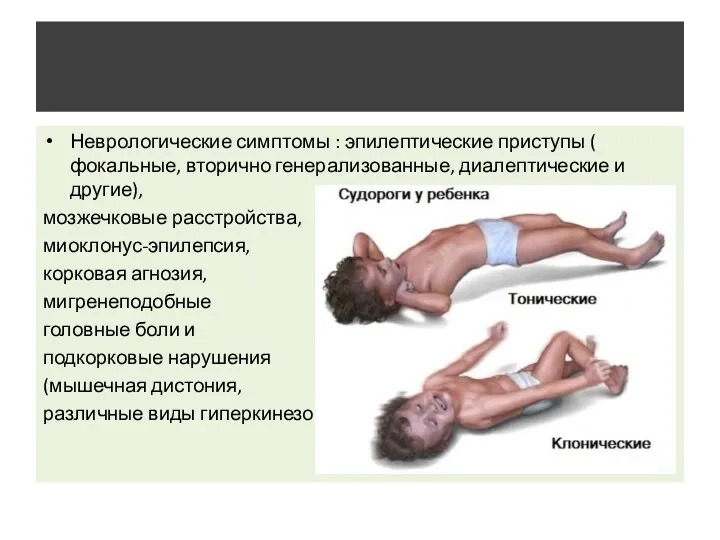
Неврологические симптомы : эпилептические приступы ( фокальные, вторично генерализованные, диалептические и
Неврологические симптомы : эпилептические приступы ( фокальные, вторично генерализованные, диалептические и
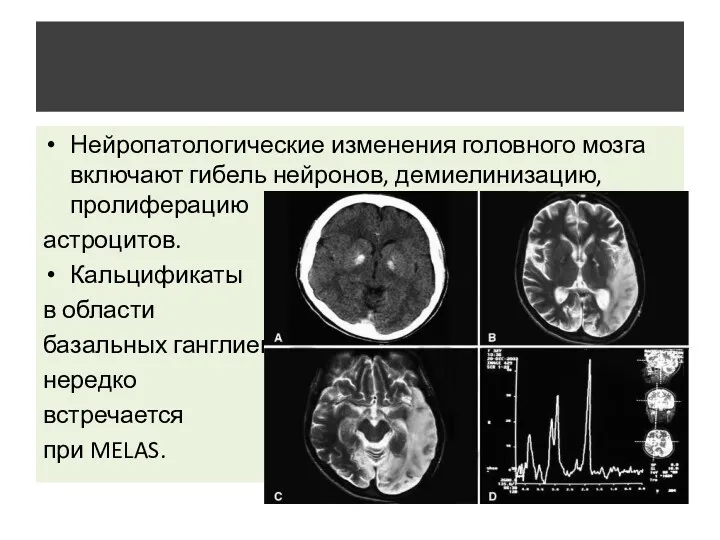
Нейропатологические изменения головного мозга включают гибель нейронов, демиелинизацию, пролиферацию
астроцитов.
Кальцификаты
Нейропатологические изменения головного мозга включают гибель нейронов, демиелинизацию, пролиферацию
астроцитов.
Кальцификаты
Мышечная слабость, нейросенсорная тугоухость являются типичными симптомами заболевания.
Эндокринопатии могут
Мышечная слабость, нейросенсорная тугоухость являются типичными симптомами заболевания.
Эндокринопатии могут

Характерен значительный уровень лактат-ацидоза в крови и спинномозговой жидкости, при
Характерен значительный уровень лактат-ацидоза в крови и спинномозговой жидкости, при

ОСОБЕННОСТИ ТЕЧЕНИЯ
Больные низкорослы
Мутация не всегда ведет в заболеванию (из-за гетероплазмии
ОСОБЕННОСТИ ТЕЧЕНИЯ
Больные низкорослы
Мутация не всегда ведет в заболеванию (из-за гетероплазмии

ДИАГНОСТИКА
Биохимические, морфологические и молекулярногенетические методы.
У всех пациентов с синдромом MELAS
ДИАГНОСТИКА
Биохимические, морфологические и молекулярногенетические методы.
У всех пациентов с синдромом MELAS
 Експертиза стійкої втрати працездатності
Експертиза стійкої втрати працездатності Дистрофии. Классификация дистрофий
Дистрофии. Классификация дистрофий Қан кету және қан түкіру кезіндегі алғашқы көмек
Қан кету және қан түкіру кезіндегі алғашқы көмек Особенности размещения приемного отделения
Особенности размещения приемного отделения Кардиальная патология при сахарном диабете
Кардиальная патология при сахарном диабете Методы лечения аномалий прикуса в периоде смешанных зубах
Методы лечения аномалий прикуса в периоде смешанных зубах Мультикиназные ингибиторы - 2
Мультикиназные ингибиторы - 2 Средства, влияющие на систему крови
Средства, влияющие на систему крови Комплексные методы лечения зубочелюстных аномалий. Виды хирургических вмешательств в возрастном аспекте
Комплексные методы лечения зубочелюстных аномалий. Виды хирургических вмешательств в возрастном аспекте лфк-при-артрозе
лфк-при-артрозе Онкологія. Організація протиракової боротьби в Україні. Етіологія і патогенез злоякісних пухлин
Онкологія. Організація протиракової боротьби в Україні. Етіологія і патогенез злоякісних пухлин Апластикалық анемиялар
Апластикалық анемиялар Хронический гломерулонефрит
Хронический гломерулонефрит Бет-жақсүйек аймағының жараларын алғашқы хирургиялық өңдеу. Ерекшеліктері. Жүргізу техникасы
Бет-жақсүйек аймағының жараларын алғашқы хирургиялық өңдеу. Ерекшеліктері. Жүргізу техникасы Бронхиалдық астма-инфекциялық аллергиялық ауру
Бронхиалдық астма-инфекциялық аллергиялық ауру Домашний диетолог. 3-фазная программа управления весом. Тест-викторина
Домашний диетолог. 3-фазная программа управления весом. Тест-викторина Коррекция нарушений развития детей: особые образовательные потребности дошкольников с ОВЗ
Коррекция нарушений развития детей: особые образовательные потребности дошкольников с ОВЗ Спортивный травматизм
Спортивный травматизм Гравидограмманың интерпретациясы
Гравидограмманың интерпретациясы Балалардағы тыныс алу жүйесін зерттеу
Балалардағы тыныс алу жүйесін зерттеу Мініінвазивна хірургія в комплексному лікуванні кіст підшлункової залози
Мініінвазивна хірургія в комплексному лікуванні кіст підшлункової залози Комплексная оценка теплового состояния среды
Комплексная оценка теплового состояния среды Процессы адаптации. Регенерация, репарация и заживление ран
Процессы адаптации. Регенерация, репарация и заживление ран Особенности питания детей школьного возраста. 8 класс
Особенности питания детей школьного возраста. 8 класс Порядок оказания скорой, в том числе скорой специализированной, медицинской помощи
Порядок оказания скорой, в том числе скорой специализированной, медицинской помощи Жүрек-қантамыр аурулары. Гипертония .Стенокардия. Инсульт миокард. Инфарктісі
Жүрек-қантамыр аурулары. Гипертония .Стенокардия. Инсульт миокард. Инфарктісі Осложнения сахарного диабета
Осложнения сахарного диабета Основные психопатологические синдромы
Основные психопатологические синдромы