- Виды заболеваний. Фенилпировиноградная олигофрения

Содержание
- 2. Фенилпировиноградная олигофрения (фенилкетонурия)
- 3. Фенилпировиноградная олигофрения,что же это такое? это распространенное наследственное заболевание, связанное с нарушением белкового обмена в организме
- 4. Виды фенилкетонурии Выделяют фенилкетонурию I, II и III типов, которые отличаются с точки зрения проявлений и
- 5. Причины возникновения фенилкетонурии -Близкородственные браки, повышают вероятность рождения ребенка больного фенилкетонурией; -Изменение (мутация) гена, локализованного на
- 6. Симптомы и признаки фенилкетонурии -Основное проявление фенилкетонурии у новорожденных это неукротимая рвота, которая иногда принимается за
- 7. Другими симптомами фенилкетонурии являются повышенная потливость со специфическим (мышиным) запахом мочи и пота, а также: Вялость
- 8. Диагностика фенилкетонурии В целях диагностики фенилкетонурии определяют уровень фенилаланина и его производных в крови, моче и
- 9. Лечение фенилкетонурии Основным и единственным на сегодняшний день методом лечения фенилкетонурии является диетотерапия. Специфическую диету при
- 10. Лекарства в лечении фенилкетонурии Препараты кальция, фосфора, железа Витамины -Препараты, улучшающие мозговое кровообращение (ноотропы – церебролизин,
- 11. Гистидинемия
- 12. Что же такое гистидинемия? Наследственная болезнь обмена веществ, обусловленная дефицитом фермента гистидин - аммиак-лиазы, характеризующаяся повышенным
- 13. Причина гистидинемии Недостаточная активность специального фермента (гистидазы), отвечающего за преобразование гистидина, который, в свою очередь, накапливается
- 14. Проявления Ухудшается общее состояние ребенка, появляются вялость, плаксивость, отказ от пищи Затем присоединяются симптомы отравления головного
- 15. Лечение Задача участкового педиатра — как можно раньше распознать заболевание (до появления осложнений со стороны нервной
- 16. Питание Продукты с низким содержанием гистидина:говяжьи почки, треска, кукурузная мука, лук, картофель, морковь, свекла, фрукты, растительное
- 17. Амавротическая идиотия
- 18. Что же такое амавротическая идиотия? Семейное прогредиентное заболевание, наблюдающееся в детском или юношеском возрасте, связанное с
- 19. Этиология и патогенез амавротической идиотии В основе заболевания лежит энхимноз, нарушение обмена липидов, ведущее к отложению
- 20. Клиника амавротической идиотии - Детская форма (Тау) -Юношеская форма (Шпильмейера-Фогта-Баттеиа) - Поздняя форма (Куфса)
- 21. Детская форма (Тау) Первые клинические признаки заболевания проявляются на 1 - 2-м году жизни. Наблюдается отставание
- 22. Юношеская форма (Шпильмейера-Фогта-Баттеиа) Заболевание возникает в возрасте 6 - 10 лет. Клинически характеризуется нарастающей неврологической симптоматикой
- 23. Поздняя форма (Куфса) Поздняя форма (Куфса) развивается у взрослых, характеризуется прогрессирующим слабоумием (энцефалопатический синдром) и неврологической
- 25. Скачать презентацию

Фенилпировиноградная олигофрения
(фенилкетонурия)
Фенилпировиноградная олигофрения
(фенилкетонурия)

Фенилпировиноградная олигофрения,что же это такое?
это распространенное наследственное заболевание, связанное с
Фенилпировиноградная олигофрения,что же это такое?
это распространенное наследственное заболевание, связанное с

Виды фенилкетонурии
Выделяют фенилкетонурию I, II и III типов, которые отличаются с
Виды фенилкетонурии
Выделяют фенилкетонурию I, II и III типов, которые отличаются с
Причины возникновения фенилкетонурии
-Близкородственные браки, повышают вероятность рождения ребенка больного фенилкетонурией;
-Изменение (мутация)
Причины возникновения фенилкетонурии
-Близкородственные браки, повышают вероятность рождения ребенка больного фенилкетонурией;
-Изменение (мутация)

Симптомы и признаки фенилкетонурии
-Основное проявление фенилкетонурии у новорожденных это неукротимая рвота, которая
Симптомы и признаки фенилкетонурии
-Основное проявление фенилкетонурии у новорожденных это неукротимая рвота, которая

Другими симптомами фенилкетонурии являются повышенная потливость со специфическим (мышиным) запахом мочи
Другими симптомами фенилкетонурии являются повышенная потливость со специфическим (мышиным) запахом мочи

Диагностика фенилкетонурии
В целях диагностики фенилкетонурии определяют уровень фенилаланина и его производных
Диагностика фенилкетонурии
В целях диагностики фенилкетонурии определяют уровень фенилаланина и его производных
Лечение фенилкетонурии
Основным и единственным на сегодняшний день методом лечения фенилкетонурии является
Лечение фенилкетонурии
Основным и единственным на сегодняшний день методом лечения фенилкетонурии является

Лекарства в лечении фенилкетонурии
Препараты кальция, фосфора, железа
Витамины
-Препараты, улучшающие мозговое кровообращение (ноотропы
Лекарства в лечении фенилкетонурии
Препараты кальция, фосфора, железа
Витамины
-Препараты, улучшающие мозговое кровообращение (ноотропы

Гистидинемия
Гистидинемия
Что же такое гистидинемия?
Наследственная болезнь обмена веществ, обусловленная дефицитом фермента гистидин - аммиак-лиазы,
Что же такое гистидинемия?
Наследственная болезнь обмена веществ, обусловленная дефицитом фермента гистидин - аммиак-лиазы,

Причина
гистидинемии
Недостаточная активность специального фермента (гистидазы), отвечающего за преобразование гистидина, который, в
Причина
гистидинемии
Недостаточная активность специального фермента (гистидазы), отвечающего за преобразование гистидина, который, в

Проявления
Ухудшается общее состояние ребенка, появляются вялость, плаксивость, отказ от пищи
Затем присоединяются
Проявления
Ухудшается общее состояние ребенка, появляются вялость, плаксивость, отказ от пищи
Затем присоединяются

Лечение
Задача участкового педиатра — как можно раньше распознать заболевание (до появления
Лечение
Задача участкового педиатра — как можно раньше распознать заболевание (до появления

Питание
Продукты с низким содержанием гистидина:говяжьи почки, треска, кукурузная мука, лук, картофель,
Питание
Продукты с низким содержанием гистидина:говяжьи почки, треска, кукурузная мука, лук, картофель,

Амавротическая идиотия
Амавротическая идиотия

Что же такое амавротическая идиотия?
Семейное прогредиентное заболевание, наблюдающееся в детском или
Что же такое амавротическая идиотия?
Семейное прогредиентное заболевание, наблюдающееся в детском или

Этиология и патогенез амавротической идиотии
В основе заболевания лежит энхимноз, нарушение обмена
Этиология и патогенез амавротической идиотии
В основе заболевания лежит энхимноз, нарушение обмена

Клиника амавротической идиотии
- Детская форма (Тау)
-Юношеская форма (Шпильмейера-Фогта-Баттеиа)
- Поздняя форма (Куфса)
Клиника амавротической идиотии
- Детская форма (Тау)
-Юношеская форма (Шпильмейера-Фогта-Баттеиа)
- Поздняя форма (Куфса)
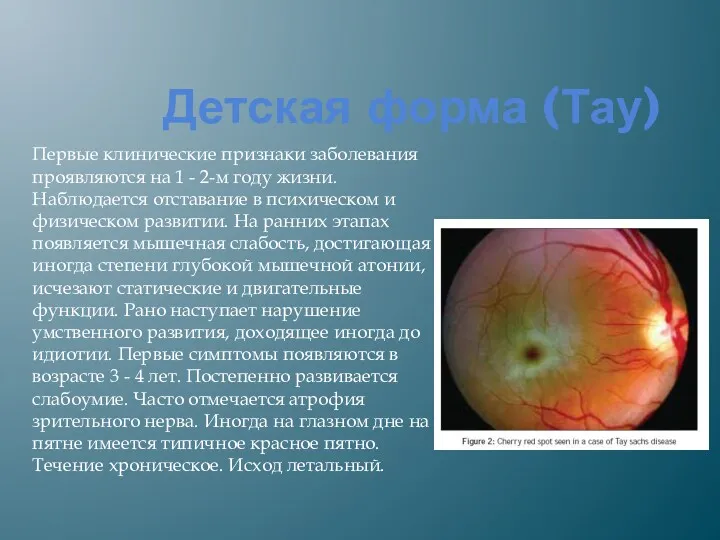
Детская форма (Тау)
Первые клинические признаки заболевания проявляются на 1 -
Детская форма (Тау)
Первые клинические признаки заболевания проявляются на 1 -

Юношеская форма (Шпильмейера-Фогта-Баттеиа)
Заболевание возникает в возрасте 6 - 10 лет. Клинически
Юношеская форма (Шпильмейера-Фогта-Баттеиа)
Заболевание возникает в возрасте 6 - 10 лет. Клинически

Поздняя форма (Куфса)
Поздняя форма (Куфса) развивается у взрослых, характеризуется прогрессирующим слабоумием
Поздняя форма (Куфса)
Поздняя форма (Куфса) развивается у взрослых, характеризуется прогрессирующим слабоумием
 Алғашқы көмек түрлері
Алғашқы көмек түрлері Ошибки диагностики и лечения внебольничных пневмоний
Ошибки диагностики и лечения внебольничных пневмоний Переход к системе непрерывного медицинского (фармацевтического) образования
Переход к системе непрерывного медицинского (фармацевтического) образования Первая медицинская помощь при острой сердечной недостаточности и инсульте
Первая медицинская помощь при острой сердечной недостаточности и инсульте Острый и хронический пиелонефрит
Острый и хронический пиелонефрит Мал шаруашылығы өнiмдерiн консервiлеу технологиясының негiздерi мен гигиенасы және оларды ветеринариялық-санитариялық сараптау
Мал шаруашылығы өнiмдерiн консервiлеу технологиясының негiздерi мен гигиенасы және оларды ветеринариялық-санитариялық сараптау Общие принципы проведения инфузионной терапиии при критических состояних. Общие принципы диагностики и ИТ эндо- и экзотоксикозов
Общие принципы проведения инфузионной терапиии при критических состояних. Общие принципы диагностики и ИТ эндо- и экзотоксикозов КОГБУЗ Яранская центральная районная больница
КОГБУЗ Яранская центральная районная больница Обезболивание на догоспитальном этапе оказания медицинской помощи
Обезболивание на догоспитальном этапе оказания медицинской помощи Принципы гигиенического нормирования вредных веществ
Принципы гигиенического нормирования вредных веществ Острая сердечная недостаточность
Острая сердечная недостаточность Лекция Медицинское обеспечение ликвидации ЧС
Лекция Медицинское обеспечение ликвидации ЧС Перинатальные инфекции новорожденных
Перинатальные инфекции новорожденных Служение больницы и учреждения
Служение больницы и учреждения Шоки-1. Патогенез шоку
Шоки-1. Патогенез шоку Қызықты клиникалық жағдай
Қызықты клиникалық жағдай Заболевания почек у детей
Заболевания почек у детей Основы трансплантологии
Основы трансплантологии Лазеры в медицине
Лазеры в медицине Отруйність спиртів та їх згубна дія на організм людини
Отруйність спиртів та їх згубна дія на організм людини Энтеробиоз. Трихинеллез
Энтеробиоз. Трихинеллез Сестринский уход при различных заболеваниях и состояниях. Лечение пациентов терапевтического профиля
Сестринский уход при различных заболеваниях и состояниях. Лечение пациентов терапевтического профиля Патофизиологический эксперимент
Патофизиологический эксперимент Рак поджелудочной железы
Рак поджелудочной железы Ситуационные задачи для проведения занятий с ординаторами. Подготовка и организация работы больницы при чрезвычайных ситуациях
Ситуационные задачи для проведения занятий с ординаторами. Подготовка и организация работы больницы при чрезвычайных ситуациях ABCDE / Einführung. Cardio-pulmonale Reanimation
ABCDE / Einführung. Cardio-pulmonale Reanimation Оказание первой помощи при несчастных случаях на производстве
Оказание первой помощи при несчастных случаях на производстве Синдром легочного инфильтрата
Синдром легочного инфильтрата