- Кортико-базальная дегенерация и прогрессирующий надъядерный паралич

Содержание
- 2. Кортико-базальная дегенерация (КБД) – редкое состояние, которое характеризуется асимметричным поражением преимущественно лобно-теменной коры, базальных ганглиев, зубчатых
- 3. Встречается с частотой 0,45 на 100 тыс., средний возраст дебюта: 63 года. Мужчины болеют несколько чаще,
- 4. Этиопатогенез: данное состояние относится к группе таупатий. Ведущий патогенетический механизм нарушение метаболизма основного белка внутренней нейрональной
- 5. Клиника: Синдром паркинсонизма в 100% случаев, в первые 3 года заболевания- ассиметричный акинетико-ригидный синдром+ постуральная неустойчивость.
- 6. Клиника: Специфическим проявлением кортикобазальной дегенерации является синдром ―чужой руки‖ (alien hand phenomenon), который наблюдается в 60%
- 7. Клиника: Апраксия, чаще идеомоторная Афазия, чаще динамическая. Глазодвигательные нарушения-апраксия открывания глаз, насильственное отведение глазных яблок. Псевдобульбарный
- 8. Критерии для постановки диагноза КБД 1.Хроническое прогрессирующее течение 2.Асимметричное начало (включая развитие афазии или апраксии) 3.Нарушение
- 9. Критерии, исключающее диагноз КБД 1. Начало с когнитивных нарушений ( иных, чем афазия или апраксия) 2.
- 10. На МРТ у больных кортикобазальной дегенерацией определяется асимметричная атрофия коры лобно-теменной области, атрофия средней части мозолистого
- 11. Лечение: Симптоматическое. У некоторых больных дофаминергические средства уменьшают симптомы паркинсонизма, но чаще эти препараты оказываются неэффективными.
- 12. Прогноз: В среднем, обездвиженность пациента наступает через 5 лет. Умирают через 5-10 лет от начала заболевания
- 13. Прогрессирующий надъядерный паралич Болезнь Стила-Ричардсона-Ольшевского: Дегенеративное заболевание головного мозга, характеризующееся скоплением тау-белка в астроцитах, нейрональных отростках
- 14. 1,39 – 6,4 на 100 тыс. населения. 4% паркинсонизма по данным патоморфологических исследований. Клинические проявления ПНП
- 15. Клиника: Одним из самых важных диагностических признаков прогрессирующего надъядерного паралича является паралич взора по вертикали, который
- 17. Диагностические критерии (the National Institute of Neurological Disorders and Stroke (NINDS) and the Society for Progressive
- 18. Вероятный диагноз: постепенно прогрессирующее заболевание с возрастом начала после 40 лет; надъядерный вертикальный паралич взора и
- 19. Критерии исключения для прогрессирующего надъядерного паралича: недавно перенесенный энцефалит; синдром «чужой руки»‖, корковые чувствительные нарушения, лобная
- 20. Подтверждающие критерии для прогрессирующего надъядерного паралича: симметричная акинезия или ригидность, более выраженная в проксимальных отделах, чем
- 21. На МРТ у больных прогрессирующим надъядерным параличом могут обнаруживаться атрофия среднего мозга и так называемый ―симптом
- 22. Лечение: Нет препаратов, способных предотвратить прогрессирования заболевания. Лечение направлено лишь на уменьшение симптомов: Дофаминовые агонисты+леводопа Трициклические
- 23. Прогноз: Продолжительность жизни от начала заболевания в среднем 5-7 лет. Через 3-5 лет возникает обездвиженность.
- 24. Лечение ндз: В настоящее время наиболее распространена симптоматическая терапия нейродегенеративных заболеваний: 1. Средства, воздействующие на специфические
- 25. 4. Коррекция экстрапирамидных нарушений при НДЗ возможна путем назначения леводопасодержащих препаратов (мадопар, наком), дофаминовых агонистов (мирапекс,
- 27. Скачать презентацию

Кортико-базальная дегенерация (КБД) – редкое состояние, которое характеризуется асимметричным поражением преимущественно
Кортико-базальная дегенерация (КБД) – редкое состояние, которое характеризуется асимметричным поражением преимущественно

Встречается с частотой 0,45 на 100 тыс., средний возраст дебюта: 63
Встречается с частотой 0,45 на 100 тыс., средний возраст дебюта: 63

Этиопатогенез:
данное состояние относится к группе таупатий.
Ведущий патогенетический механизм
Этиопатогенез:
данное состояние относится к группе таупатий.
Ведущий патогенетический механизм

Клиника:
Синдром паркинсонизма в 100% случаев, в первые 3 года заболевания- ассиметричный
Клиника:
Синдром паркинсонизма в 100% случаев, в первые 3 года заболевания- ассиметричный

Клиника:
Специфическим проявлением кортикобазальной дегенерации является синдром ―чужой руки‖ (alien hand phenomenon),
Клиника:
Специфическим проявлением кортикобазальной дегенерации является синдром ―чужой руки‖ (alien hand phenomenon),

Клиника:
Апраксия, чаще идеомоторная
Афазия, чаще динамическая.
Глазодвигательные нарушения-апраксия открывания глаз, насильственное отведение глазных
Клиника:
Апраксия, чаще идеомоторная
Афазия, чаще динамическая.
Глазодвигательные нарушения-апраксия открывания глаз, насильственное отведение глазных

Критерии для постановки диагноза КБД
1.Хроническое прогрессирующее течение
2.Асимметричное начало (включая развитие
Критерии для постановки диагноза КБД
1.Хроническое прогрессирующее течение
2.Асимметричное начало (включая развитие

Критерии, исключающее диагноз КБД
1. Начало с когнитивных нарушений ( иных, чем
Критерии, исключающее диагноз КБД
1. Начало с когнитивных нарушений ( иных, чем
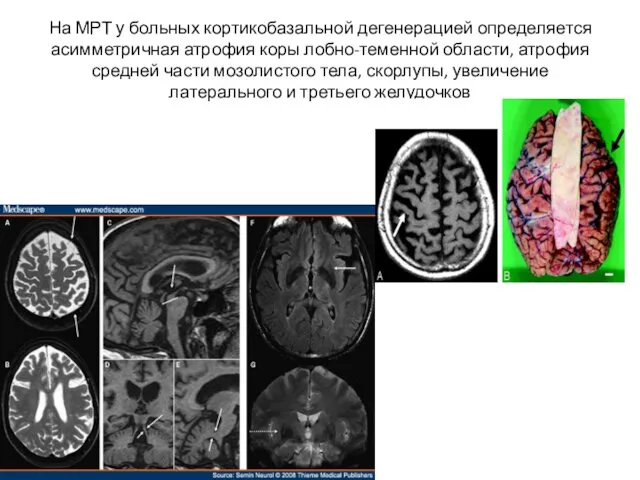
На МРТ у больных кортикобазальной дегенерацией определяется асимметричная атрофия коры лобно-теменной
На МРТ у больных кортикобазальной дегенерацией определяется асимметричная атрофия коры лобно-теменной

Лечение:
Симптоматическое. У некоторых больных дофаминергические средства уменьшают симптомы паркинсонизма, но чаще
Лечение:
Симптоматическое. У некоторых больных дофаминергические средства уменьшают симптомы паркинсонизма, но чаще

Прогноз:
В среднем, обездвиженность пациента наступает через 5 лет.
Умирают через 5-10 лет
Прогноз:
В среднем, обездвиженность пациента наступает через 5 лет.
Умирают через 5-10 лет

Прогрессирующий надъядерный паралич
Болезнь Стила-Ричардсона-Ольшевского:
Дегенеративное заболевание головного мозга, характеризующееся скоплением
Прогрессирующий надъядерный паралич
Болезнь Стила-Ричардсона-Ольшевского:
Дегенеративное заболевание головного мозга, характеризующееся скоплением

1,39 – 6,4 на 100 тыс. населения. 4% паркинсонизма по данным
1,39 – 6,4 на 100 тыс. населения. 4% паркинсонизма по данным

Клиника:
Одним из самых важных диагностических признаков прогрессирующего надъядерного паралича является паралич
Клиника:
Одним из самых важных диагностических признаков прогрессирующего надъядерного паралича является паралич


Диагностические критерии
(the National Institute of Neurological Disorders and
Stroke (NINDS) and
Диагностические критерии (the National Institute of Neurological Disorders and Stroke (NINDS) and

Вероятный диагноз:
постепенно прогрессирующее заболевание с возрастом начала после 40 лет;
надъядерный
Вероятный диагноз:
постепенно прогрессирующее заболевание с возрастом начала после 40 лет;
надъядерный

Критерии исключения для прогрессирующего надъядерного паралича:
недавно перенесенный энцефалит;
синдром «чужой руки»‖,
Критерии исключения для прогрессирующего надъядерного паралича:
недавно перенесенный энцефалит;
синдром «чужой руки»‖,

Подтверждающие критерии для прогрессирующего надъядерного паралича:
симметричная акинезия или ригидность, более выраженная
Подтверждающие критерии для прогрессирующего надъядерного паралича:
симметричная акинезия или ригидность, более выраженная
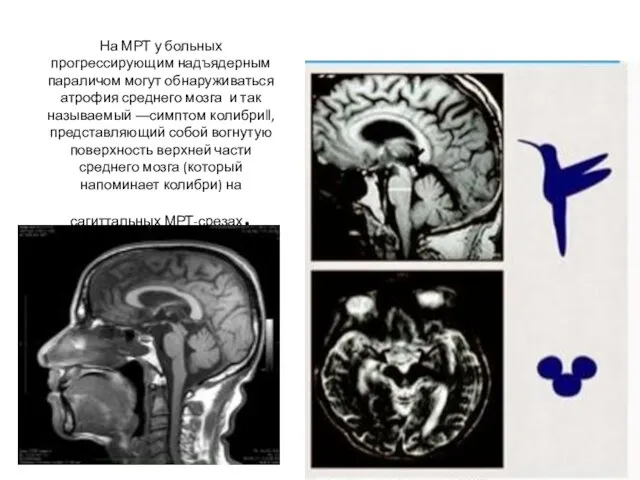
На МРТ у больных прогрессирующим надъядерным параличом могут обнаруживаться атрофия среднего
На МРТ у больных прогрессирующим надъядерным параличом могут обнаруживаться атрофия среднего

Лечение:
Нет препаратов, способных предотвратить прогрессирования заболевания. Лечение направлено лишь на уменьшение
Лечение:
Нет препаратов, способных предотвратить прогрессирования заболевания. Лечение направлено лишь на уменьшение

Прогноз:
Продолжительность жизни от начала заболевания в среднем 5-7 лет.
Через 3-5 лет
Прогноз:
Продолжительность жизни от начала заболевания в среднем 5-7 лет.
Через 3-5 лет

Лечение ндз:
В настоящее время наиболее распространена симптоматическая терапия нейродегенеративных заболеваний:
1. Средства, воздействующие
Лечение ндз:
В настоящее время наиболее распространена симптоматическая терапия нейродегенеративных заболеваний:
1. Средства, воздействующие

4. Коррекция экстрапирамидных нарушений при НДЗ возможна путем назначения леводопасодержащих препаратов
4. Коррекция экстрапирамидных нарушений при НДЗ возможна путем назначения леводопасодержащих препаратов
 Адміністративне право. Курс лекцій
Адміністративне право. Курс лекцій Смотр знаний Генетическая связь между основными классами неорганических соединений.
Смотр знаний Генетическая связь между основными классами неорганических соединений. Экзистенциализм
Экзистенциализм Портфолио класса
Портфолио класса Oil and gas industry segments. Part 1
Oil and gas industry segments. Part 1 Технологии презентаций
Технологии презентаций Тригонометрические выражения
Тригонометрические выражения Урок-игра Углеводороды
Урок-игра Углеводороды Урок-38
Урок-38 Евгений Онегин. Автор и его образ. Лирические отступления
Евгений Онегин. Автор и его образ. Лирические отступления Задачи на проценты. Подготовка к ЕГЭ. Задания В1
Задачи на проценты. Подготовка к ЕГЭ. Задания В1 Описание Сморгонского района
Описание Сморгонского района Обыкновенные и десятичные дроби
Обыкновенные и десятичные дроби Кубань - Любимый уголок земли.
Кубань - Любимый уголок земли. Структура строительного комплекса. Типы предприятий строительных изделий и конструкций, производственная структура предприятий
Структура строительного комплекса. Типы предприятий строительных изделий и конструкций, производственная структура предприятий What day is it today?
What day is it today? Медико-биологические и социальные основы здоровья
Медико-биологические и социальные основы здоровья Материалы внешней отделки в архитектуре музеев
Материалы внешней отделки в архитектуре музеев Основные изменения законодательства Российской Федерации по налогу на имущество физических лиц (2015)
Основные изменения законодательства Российской Федерации по налогу на имущество физических лиц (2015) Конкурс чтецов
Конкурс чтецов Конструкции плоских перекрытий. Классификация
Конструкции плоских перекрытий. Классификация Хирургиялық деонтология
Хирургиялық деонтология Органы растений
Органы растений Стабилизация нефти. Этапы сбора и подготовки нефти на промыслах
Стабилизация нефти. Этапы сбора и подготовки нефти на промыслах Военные реформы в истории Российского государства: опыт и уроки
Военные реформы в истории Российского государства: опыт и уроки Снегири на ветках.
Снегири на ветках. Агрессивные дети
Агрессивные дети Рабочая тетрадь
Рабочая тетрадь