- Методика выделения ДНК, оценка качества выделения

Содержание
- 2. Этапы выделения ДНК из клеточного образца 0. Гомогенизация образца. 1. Лизис клеток. – SDS (SLS, додецилсульфат
- 3. Этап 0. Гомогенизация образца ткани Этап требуется в случае необходимости выделения ДНК из материала, содержащего прочные
- 4. Этап 1. Лизис клеток Разрушение клеточной оболочки – клеточной стенки (если есть) и мембран 1. Физический
- 5. Этап 2. Удаление примесей Разрушение “ненужных” нуклеиновых кислот специфическими нуклеазами (напр., РНКаза) Разрушение белков протеазами (напр.,
- 6. Этап 3. Преципитация ДНК 1. Осаждение ДНК из раствора: соль (NaCl, AcNa) + концентрированный спирт (изопропанол
- 7. Этап 4. Растворение ДНК Зачастую в качестве буфера для растворения (или элюции) ДНК используют TE-буфер (Tris/EDTA-буфер)
- 8. Сохранность выделенной ДНК в TE-буфере +4ºC: недели -20ºC: месяцы -80ºC: годы
- 9. Выделение ДНК фенол-хлороформным методом
- 10. Разделение ДНК и примесей по градиенту плотности в фенол-хлороформном методе
- 11. Варианты фенольной экстракции при выделении ДНК или РНК Для выделения РНК используют реагент, содержащий GITC и
- 12. Преимущества и недостатки метода фенол-хлороформной экстракции Преимущества: подходит для выделения из разных материалов является “золотым стандартом”
- 13. Метод выделения ДНК простым осаждением
- 14. Преимущества и недостатки метода выделения простым осаждением Преимущества: по качеству выделения сопоставим с ФХЭ не требует
- 15. Выделение ДНК на частицах силикагеля
- 16. Выделение ДНК на колонках
- 17. Связывание ДНК на поверхности колонки
- 18. Выделение ДНК с использованием магнитных частиц
- 19. Варианты магнитных частиц
- 20. Магнитный штатив
- 21. Преимущества и недостатки метода выделения на сорбенте (частицы силикагеля, колонки, магнитные частицы) Преимущества: сравнительно быстрый метод
- 22. Параметры оценки качества выделения ДНК 1. Количество выделенной ДНК (концентрация) – спектрофотометр – флюориметр 2. Качество
- 23. Спектрофотометр Спектрофотометр измеряет отношение интенсивностей падающего на вещество (раствор) и прошедшего через него потоков света определённой
- 24. Спектрофотометрия: оценка концентрации ДНК C (мкг/мл) = Aλ * K C – концентрация вещества (ДНК) в
- 25. Оценка содержания примесей и чистоты ДНК Содержание примесей оценивается по величине отношения поглощения света при разных
- 26. Флюориметрическое (спектрофлюориметрическое) определение концентрации ДНК К исследуемому образцу добавляют специфический флюорохром и определяют интенсивность флюоресценции при
- 27. Преимущества и недостатки флюориметрии по сравнению со спектрофотометрией Преимущества: высокая селективность (за счёт специфичного связывания флюорохрома)
- 28. Принцип гель-электрофореза НК
- 29. Электрофорез: 1. Внесение образца ДНК в лунку геля Пластину агарозного геля кладут в камеру для фореза.
- 30. Электрофорез: 2. Запуск электрофореза Камеру для фореза закрывают крышкой, устанавливают режим (напряжение, время) электрофореза.
- 31. Электрофорез: 3. Просмотр в трансиллюминаторе Гель перемещают в трансиллюминатор, где происходит облучение геля ультрафиолетом. Результат оценивают
- 33. Скачать презентацию

Этапы выделения ДНК из клеточного образца
0. Гомогенизация образца.
1. Лизис клеток.
– SDS
Этапы выделения ДНК из клеточного образца
0. Гомогенизация образца.
1. Лизис клеток.
– SDS

Этап 0. Гомогенизация образца ткани
Этап требуется в случае необходимости выделения ДНК
Этап 0. Гомогенизация образца ткани
Этап требуется в случае необходимости выделения ДНК

Этап 1. Лизис клеток
Разрушение клеточной оболочки – клеточной стенки (если есть)
Этап 1. Лизис клеток
Разрушение клеточной оболочки – клеточной стенки (если есть)

Этап 2. Удаление примесей
Разрушение “ненужных” нуклеиновых кислот специфическими нуклеазами (напр., РНКаза)
Разрушение
Этап 2. Удаление примесей
Разрушение “ненужных” нуклеиновых кислот специфическими нуклеазами (напр., РНКаза)
Разрушение

Этап 3. Преципитация ДНК
1. Осаждение ДНК из раствора:
соль (NaCl, AcNa)
Этап 3. Преципитация ДНК
1. Осаждение ДНК из раствора:
соль (NaCl, AcNa)
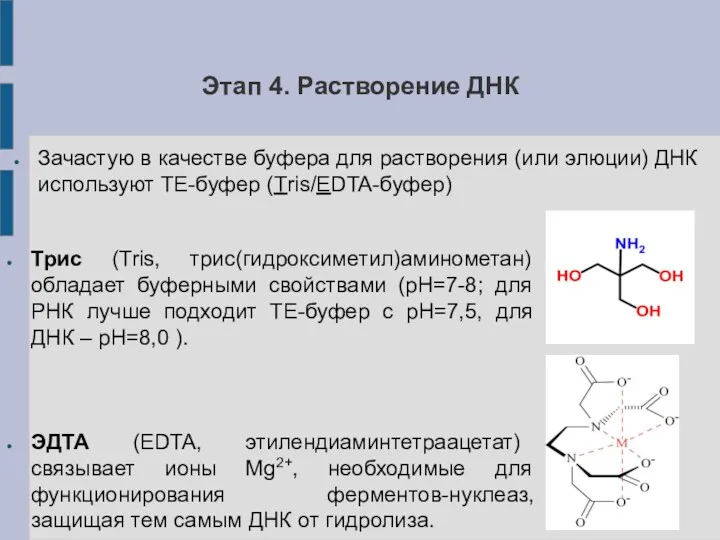
Этап 4. Растворение ДНК
Зачастую в качестве буфера для растворения (или элюции)
Этап 4. Растворение ДНК
Зачастую в качестве буфера для растворения (или элюции)

Сохранность выделенной ДНК в TE-буфере
+4ºC: недели
-20ºC: месяцы
-80ºC: годы
Сохранность выделенной ДНК в TE-буфере
+4ºC: недели
-20ºC: месяцы
-80ºC: годы
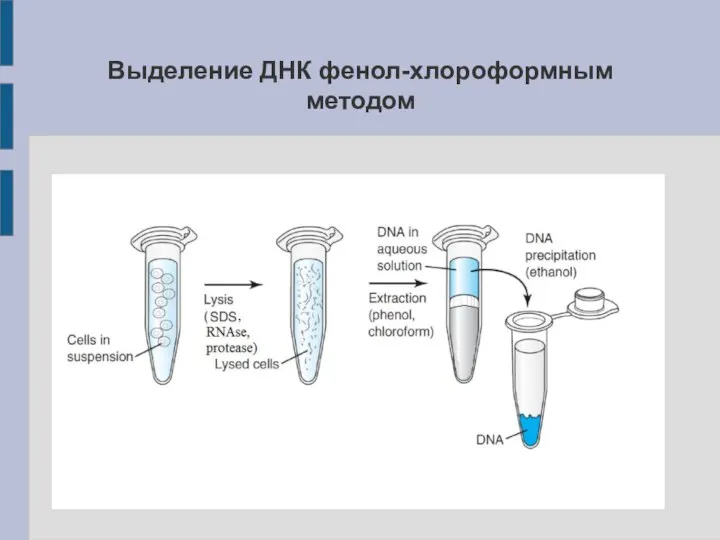
Выделение ДНК фенол-хлороформным методом
Выделение ДНК фенол-хлороформным методом
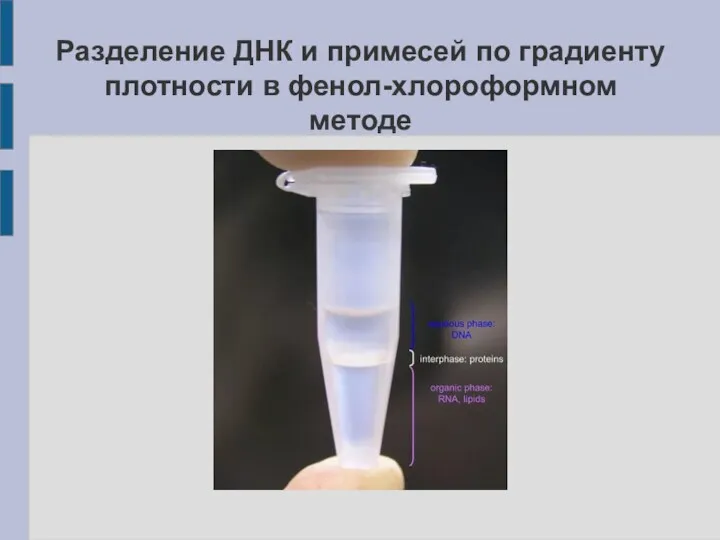
Разделение ДНК и примесей по градиенту плотности в фенол-хлороформном методе
Разделение ДНК и примесей по градиенту плотности в фенол-хлороформном методе
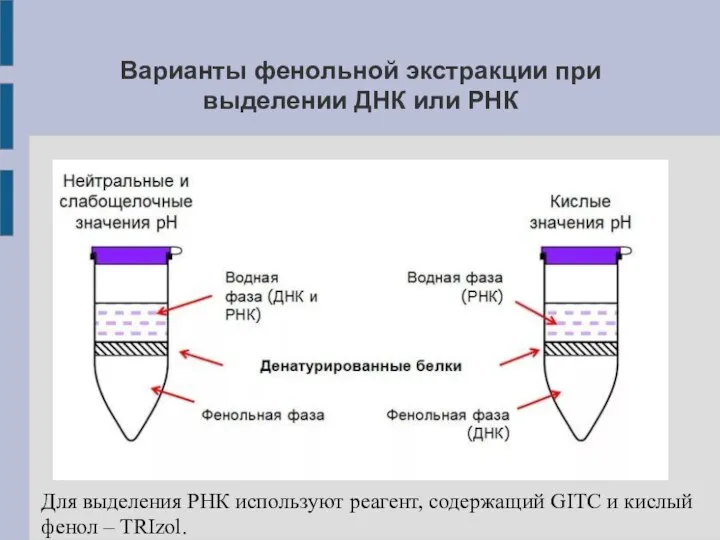
Варианты фенольной экстракции при выделении ДНК или РНК
Для выделения РНК используют
Варианты фенольной экстракции при выделении ДНК или РНК
Для выделения РНК используют

Преимущества и недостатки метода фенол-хлороформной экстракции
Преимущества:
подходит для выделения из разных материалов
является
Преимущества и недостатки метода фенол-хлороформной экстракции
Преимущества:
подходит для выделения из разных материалов
является
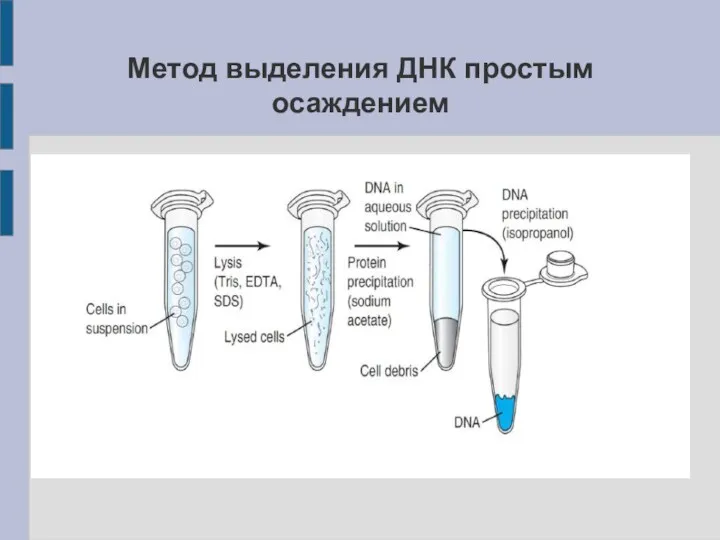
Метод выделения ДНК простым осаждением
Метод выделения ДНК простым осаждением

Преимущества и недостатки метода выделения простым осаждением
Преимущества:
по качеству выделения сопоставим с
Преимущества и недостатки метода выделения простым осаждением
Преимущества:
по качеству выделения сопоставим с
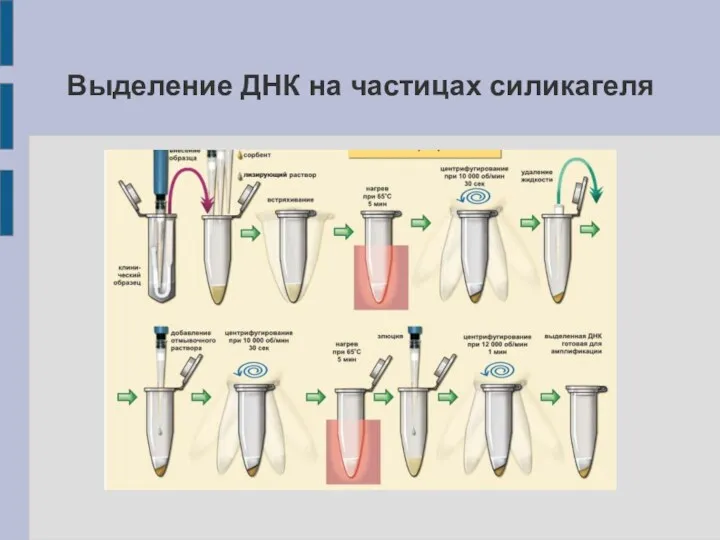
Выделение ДНК на частицах силикагеля
Выделение ДНК на частицах силикагеля
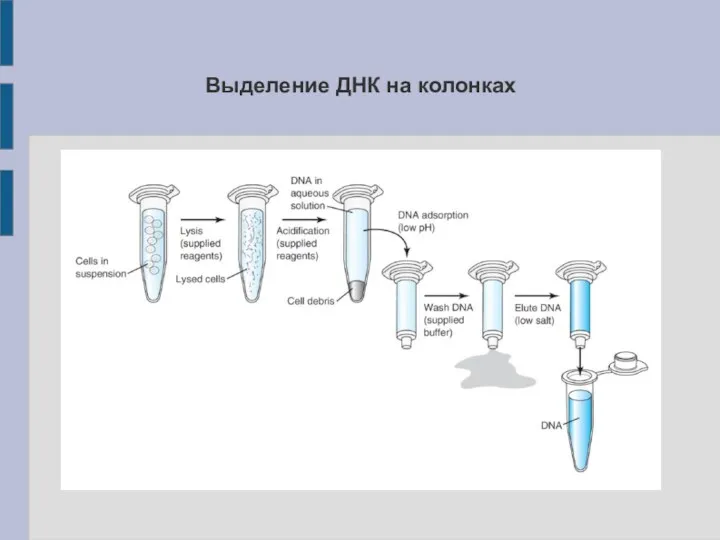
Выделение ДНК на колонках
Выделение ДНК на колонках
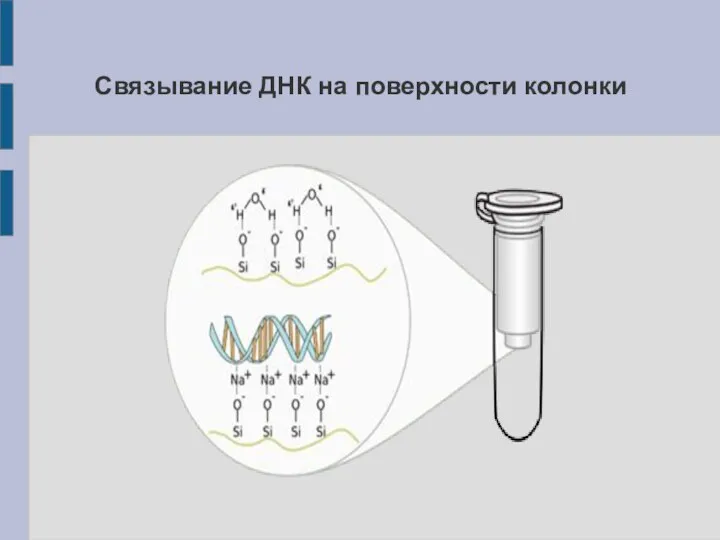
Связывание ДНК на поверхности колонки
Связывание ДНК на поверхности колонки
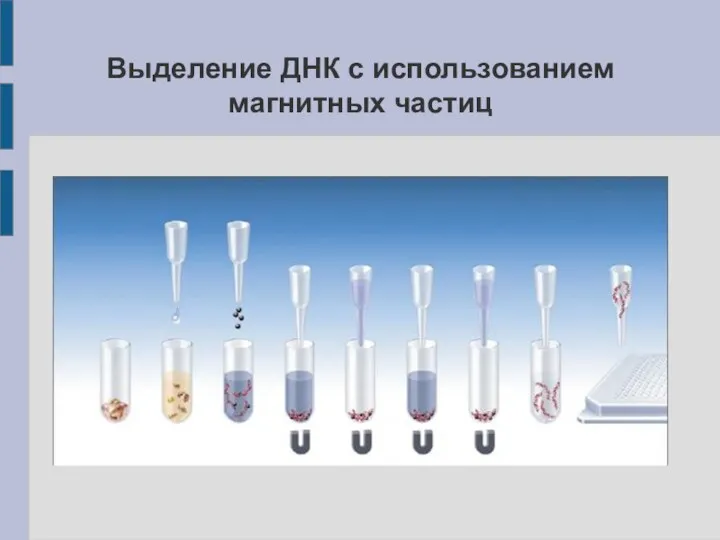
Выделение ДНК с использованием магнитных частиц
Выделение ДНК с использованием магнитных частиц
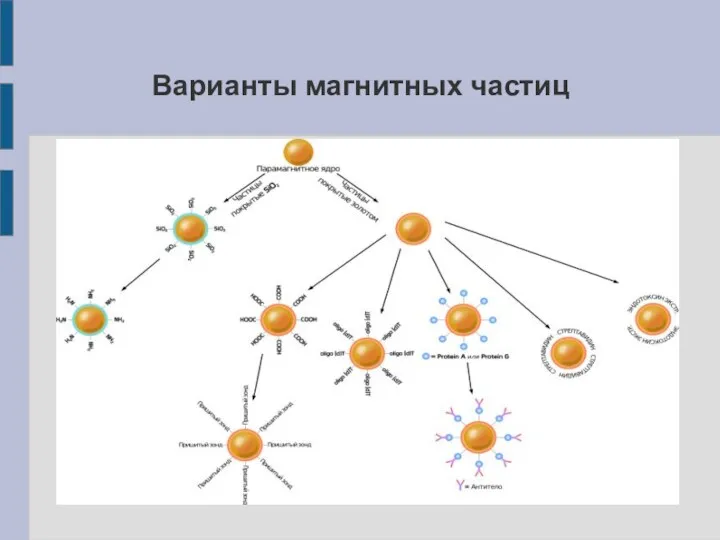
Варианты магнитных частиц
Варианты магнитных частиц

Магнитный штатив
Магнитный штатив

Преимущества и недостатки метода выделения на сорбенте (частицы силикагеля, колонки, магнитные
Преимущества и недостатки метода выделения на сорбенте (частицы силикагеля, колонки, магнитные

Параметры оценки качества выделения ДНК
1. Количество выделенной ДНК (концентрация)
– спектрофотометр
– флюориметр
2.
Параметры оценки качества выделения ДНК
1. Количество выделенной ДНК (концентрация)
– спектрофотометр
– флюориметр
2.

Спектрофотометр
Спектрофотометр измеряет отношение интенсивностей падающего на вещество (раствор) и прошедшего через
Спектрофотометр
Спектрофотометр измеряет отношение интенсивностей падающего на вещество (раствор) и прошедшего через

Спектрофотометрия: оценка концентрации ДНК
C (мкг/мл) = Aλ * K
C – концентрация
Спектрофотометрия: оценка концентрации ДНК
C (мкг/мл) = Aλ * K
C – концентрация

Оценка содержания примесей и чистоты ДНК
Содержание примесей оценивается по величине отношения
Оценка содержания примесей и чистоты ДНК
Содержание примесей оценивается по величине отношения

Флюориметрическое (спектрофлюориметрическое) определение концентрации ДНК
К исследуемому образцу добавляют специфический флюорохром и
Флюориметрическое (спектрофлюориметрическое) определение концентрации ДНК
К исследуемому образцу добавляют специфический флюорохром и

Преимущества и недостатки флюориметрии по сравнению со спектрофотометрией
Преимущества:
высокая селективность (за счёт
Преимущества и недостатки флюориметрии по сравнению со спектрофотометрией
Преимущества:
высокая селективность (за счёт
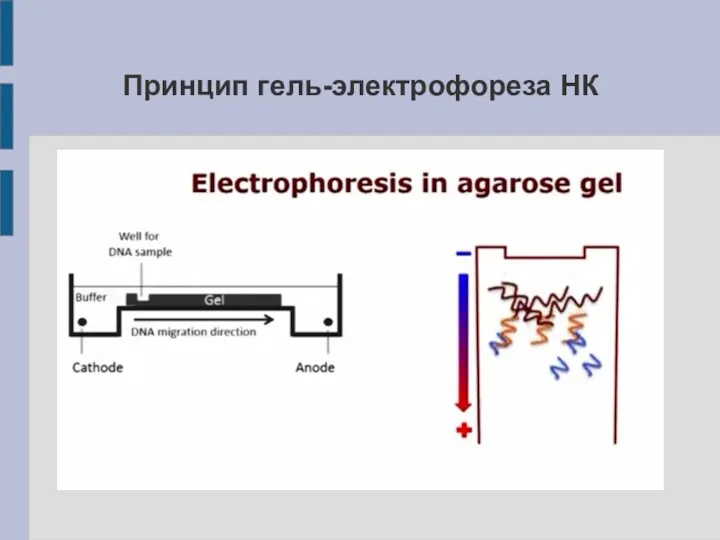
Принцип гель-электрофореза НК
Принцип гель-электрофореза НК

Электрофорез: 1. Внесение образца ДНК в лунку геля
Пластину агарозного геля кладут
Электрофорез: 1. Внесение образца ДНК в лунку геля
Пластину агарозного геля кладут

Электрофорез: 2. Запуск электрофореза
Камеру для фореза закрывают крышкой, устанавливают режим (напряжение,
Электрофорез: 2. Запуск электрофореза
Камеру для фореза закрывают крышкой, устанавливают режим (напряжение,

Электрофорез: 3. Просмотр в трансиллюминаторе
Гель перемещают в трансиллюминатор, где происходит облучение
Электрофорез: 3. Просмотр в трансиллюминаторе
Гель перемещают в трансиллюминатор, где происходит облучение
 Методы определения параметров гумусового состояния почв
Методы определения параметров гумусового состояния почв Углеводы. Моносахариды. (Лекция 13)
Углеводы. Моносахариды. (Лекция 13) Методы селекции
Методы селекции Рёбра. Анатомия. Физиология
Рёбра. Анатомия. Физиология Корсак
Корсак Микробиология - наука о микроорганизмах. История становления
Микробиология - наука о микроорганизмах. История становления Протопласты растительных клеток, как объекты биологического конструировании
Протопласты растительных клеток, как объекты биологического конструировании Микробиология
Микробиология Регуляція роботи серця
Регуляція роботи серця Проводящие пути ЦНС
Проводящие пути ЦНС The methods for the preparation of permanent stains. the structure and the working principle of the fermenter
The methods for the preparation of permanent stains. the structure and the working principle of the fermenter Плазмиды
Плазмиды Адаптации организмов к условиям обитания
Адаптации организмов к условиям обитания Вода - основной источник жизни и здоровья
Вода - основной источник жизни и здоровья Древнейшие люди
Древнейшие люди Световая микроскопия. Занятие 1
Световая микроскопия. Занятие 1 Строение эукариотической клетки
Строение эукариотической клетки Цитологический атлас
Цитологический атлас Гости из далёких стран
Гости из далёких стран Растения и окружающая среда
Растения и окружающая среда Перелетные птицы весной
Перелетные птицы весной Серая ворона
Серая ворона Микроорганизмы – инструменты научных исследований
Микроорганизмы – инструменты научных исследований Семейство сложноцветные (астровые)
Семейство сложноцветные (астровые) Капуста
Капуста Кодирование информации в нервной системе
Кодирование информации в нервной системе Дәрілік препараттардың синтезі мен олардың классификациясы
Дәрілік препараттардың синтезі мен олардың классификациясы Зимующие птицы
Зимующие птицы