- Электронная спектроскопия

Содержание
- 2. Спектр - в физике — распределение значений физической величины. Обычно под спектром подразумевается электромагнитный спектр —
- 4. При исследовании строения химических соединений, наибольшую информацию можно получить при изучении взаимодействия вещества с электромагнитным излучением.
- 5. Различные виды излучения характеризуются различной энергией - энергия излучения, поглощаемая молекулой, при взаимодействии с ней электрической
- 6. Условия поглощения излучения: энергия излучения должна совпадать с разностью энергий квантованных уровней ∆Е, соответствующих различным состояниям
- 7. Е*>Е0 происходит поглощение света – спектры поглощения Е0 > Е* происходит излучение энергии - эмиссионные спектры
- 9. Излучение можно охарактеризовать: длиной волны λ волновым числом υ частотой ΰ Эти параметры связаны между собой
- 10. Электромагнитный спектр
- 11. Электромагнитный спектр
- 12. Чем выше энергия излучения, тем меньше длинна волны и больше частота и волновое число. Энергия излучения
- 13. Электронные спектры поглощения возникают в результате переходов электронов из основного в возбужденное состояние. Для возбуждения электрона
- 14. Электронные спектры относятся к УФ, видимой и ближней ИК области электромагнитного спектра (120 – 1000 нм).
- 16. Законы поглощения света Для оптических спектров имеются общие законы поглощения излучения, дающие соотношение между величиной поглощения
- 17. k = 0,434k/ - закон Бугера - Ламберта Первый закон выражает зависимость между интенсивностью прошедшего излучения
- 18. Второй закон выражает связь между интенсивностью прошедшего излучения и концентрацией поглощающего вещества в растворе: поток параллельных
- 20. Способы изображения электронных спектров Электронные спектры поглощения записываются в виде зависимости двух переменных величин – фактора
- 21. Природа электронных спектров При обработке молекулярных спектров пользуются приближением Борна – Оппенгеймера: полная энергия системы рассматривается
- 22. Энергетические состояния двухатомной молекулы. ν0, ν1, ν0/, ν1/ - обозначают колебательные уровни одного колебания в основном
- 23. Колебательные и электронные энергетические уровни двухатомной молекулы Для каждого электронного состояния характерна своя функция потенциальной энергии.
- 24. Кривые Морзе для основного и возбужденного состояний двухатомной молекулы; штриховыми линиями показаны вероятностные колебательные функции ψvib
- 25. Основные положения электронный спектроскопии Принцип Франка – Кондона. За то очень короткое время, которое необходимо для
- 27. 2. Правило отбора Общего правила отбора, которое налагало бы ограничения на изменения в колебательном состоянии, сопровождающие
- 28. Относительная интенсивность различных колебательных подполос зависит от колебательной волновой функции для различных уровней. Переход выгоден, если
- 29. 3. Требование по симметрии электронных переходов. Приведенное выше обсуждение касалось двухатомной молекулы, но основные его положения
- 30. По аналогии с теорией молекулярных орбиталей: переход молекулы из основного состояния в возбужденное соответствует переходу валентного
- 31. Последовательность энергетических уровней электронов в молекуле органических соединений:
- 32. σ → σ* - вакуумная УФ - 100 - 200 нм, (предельные углеводороды) π → π*
- 33. Элементы симметрии – оси, плоскости, центры инверсии Операцией симметрии молекулярной системы называют такое ее движение относительно
- 34. Типы симметрии состояний молекулы: А – симметрия относительно главной поворотной оси В – антисимметрия относительно главной
- 35. Электронные переходы в карбонильной группе формальдегида Относительные энергии молекулярных орбиталей карбонильной группы H2CO Последовательность молекулярных орбиталей,
- 36. Формы молекулярных орбиталей формальдегида
- 37. Для нахождения симметрии МО определяем полную симметрию молекулы, которая в данном случае относится к С2v. Далее
- 38. В основном состоянии на каждой МО находится по два электрона и оно всегда является полносимметричным А1.
- 39. Зная симметрию орбиталей в основном состоянии можно определить симметрию возбужденного состояния. Тип симметрии состояния является произведением
- 41. В состоянии, возникающем при переходе n→π*, по одному неспаренному электрону находится на орбитали n симметрии b2
- 42. Интенсивность электронных переходов. Сила осциллятора υ – волновое число (см-1) - интегральная интенсивность, которая измеряется площадью
- 43. Величина f безразмерная для полностью разрешенного перехода f = 1 Интенсивность электронных переходов в спектрах поглощения
- 44. Сила осциллятора может быть вычислена по формуле: - вероятность перехода между двумя состояниями m и n
- 45. Момент перехода связан с волновыми функциями начального и конечного состояний соотношением: - интеграл момента перехода; ψm
- 46. не обращается в ноль только в том случае, если подынтегральное выражение полносимметрично, а для этого необходимо,
- 47. Разберем на примере электронных переходов в формальдегиде. 1) π→π* переход. Волновая функция возбужденного состояния может быть
- 48. 2) n → π* А1 → А2. Из таблицы характеров видно, что ни одна из компонент
- 49. Правила отбора электронных переходов 1. По мультиплетности. Запрещены переходы между состояниями различной мультиплетности. 2. По симметрии.
- 50. Перечисленные правила отбора выведены из рассмотрения только электронных волновых функций, без учета колебательных и вращательных волновых
- 51. Хромофоры и ауксохромы Поглощение вещества в ближнем УФ и видимой области связано с возбуждением π→π* или
- 52. 1. Очень интенсивные полосы с ε > 103 соответствующие π→π* переходам, типичные для конъюгированных (сопряженных) систем,
- 53. Замещение или изменение структуры органический соединений вызывает изменение длины волны и интенсивности полосы поглощения. Смещения полос
- 54. Влияние растворителя на электронные спектры веществ Структура и положение полос поглощения зависят от природы растворителя. Полярные
- 56. Замена полярного растворителя на инертный позволяет установить некоторые структурные особенности органических молекул. В частности, такая замена
- 57. Замена растворителя может привести к изменению таутомерного равновесия. При этом если таутомеры содержат разные хромофоры, то
- 58. Использование в качестве растворителе спиртовых и водных растворов кислот и щелочей часто оказывается полезным для структурного
- 59. Для обнаружения хромофоров, содержащих основный азот (например хромофор С=N в алифатических азометинах) снимают спектр в кислотном
- 60. Электронные спектры поглощения отдельных классов органических соединений Насыщенные соединения В насыщенных углеводородах (парафинах и циклопарафинах), содержащие
- 61. Ненасыщенные соединения Непредельные углеводороды с изолированными двойными связями имеют интенсивную полосу поглощения, обусловленную π→π* переходом, в
- 65. Карбонильные соединения Карбонильные соединения содержат гетероатомы, связанные кратной связью, в таких группах возможны три типа электронных
- 68. Непредельные карбонильные соединения. Сопряжение кратной связи с карбонильной группой вызывает длинноволновое смещение n→π*, π→π* переходов по
- 70. Тиокарбонильные соединения. Тиокарбонильная группа по своему поглощению аналогична карбонильной, однако ее полосы расположены в более длинноволновой
- 71. Бензол и его производные Спектр поглощения бензола в гептане Бензол имеет три полосы поглощения, связанные с
- 72. Алкильные заместители и галогены приводят к небольшому смещению в длинноволновую область и увеличению интенсивности полос при
- 73. При введении в бензольное кольцо полярных групп, содержащих неподеленные электронные пары (OH, OR, NH2, NR2), происходит
- 74. При блокировании неподеленных электронов, например, в случае солеобразования по аминогруппе спектр поглощение приближается к спектру алкильных
- 75. Заместители, содержащие кратные связи, вызывают увеличение интенсивности полос поглощения и смещение их в длинноволновую область спектра.
- 76. Если заместителем является карбонильная группа, в спектре могут наблюдаться полосы перехода n→π*. Они хорошо видны в
- 77. При накоплении сопряженных кратных связей в заместителе появляется широкая интенсивная полоса поглощения, характеризующая общую сопряженную систему
- 78. При наличии двух заместителей в бензольном кольце спектр соединения зависит от природы заместителей и от их
- 80. Электронные спектры комплексов переходных металлов Электронные спектры комплексов переходных металлов интерпретируют с помощью теории кристаллического поля
- 84. Td
- 85. D4h (MX4Y2)
- 86. В зависимости величины параметра ∆ и межэлектронного взаимодействия зависит распределение электронов по орбиталям. Увеличение параметра ∆
- 87. Порядок величины 10Dq правильно передается энергией самого длинноволнового спектрального перехода в комплексах переходных металлов. Природа самой
- 88. Расщепление энергетических состояний для dn-конфигураций центральных ионов Определенное энергетическое состояние атома называют атомным термом. Любое обозначение
- 89. В октаэдрическом и тетраэдрическом, а также кубических электростатических полях лигандов термы центрального иона независимо от мультиплетности
- 91. Диаграммы Оргела Диаграмма Оргела для высокоспиновых d1, d9, d4, d6 - комплексов
- 92. Диаграмма Оргела для высокоспиновых d2, d8, d3, d7 - комплексов
- 93. Диаграмма Танабе-Сугано
- 96. Нефелоуксетический ряд F->H2O>urea>NH3> en~C2O42-> NCS-> Cl-~CN- >Br-> S2- ~I-.
- 98. 3А2g→3T2g (H2O-8500; NH3-10500 см-1) 3А2g→3T1g(F) (H2O-15400; NH3-17500 см-1) 3А2g→3T1g(P) (H2O-26000; NH3-28200 см-1) 1 – Ni(NH3)62+, 2
- 99. Уравнения Танабе – Сугано для Ni2+ в октаэдрическом поле E (3Т2g) = -2Dq E (3A2g) =
- 100. Построение диаграммы Оргела 1. Энергию основного состояния центрального иона (энергию терма) принимаем за ноль. 2. Находим
- 101. Построение диаграммы Оргела для октаэдрических комплексов Ni2+ Энергию основного состояния Ni2+ т.е энергию терма 3F, принимаем
- 102. Вычисляем по уравнениям энергии компонент расщепления терма 3F: 3A2g, 3T2g, 3T1g , а также энергию 3T1g
- 104. Эффект Яна-Тейлера
- 107. [Co(H2O)6]2+ [CoCl4]2+
- 108. Комплексы никеля: 1 – D2d, 2 – Oh, 3 – Td, 4 - D4h
- 109. Методы определения электрических дипольных моментов молекул Диэлектрическая проницаемость и электрическая поляризация диэлектрика. Дипольный момент молекулы Диэлектрики
- 110. Полярным диэлектриком называется такой диэлектрик, молекулы (атомы) которого имеют электроны, расположенные несимметрично относительно своих ядер (H2O,
- 111. Если неполярный диэлектрик поместить в электрическое поле конденсатора, то происходит деформация электронных оболочек в атомах (молекулах)
- 112. В полярных диэлектриках молекулы представляют собой электрические диполи, которые в отсутствии электрического поля ориентированы хаотически (суммарный
- 113. Заполнение пространства между пластинами конденсатора диэлектриком приводит к уменьшению напряженности поля в ε раз. εs =
- 114. Уменьшение напряженности электрического поля в конденсаторе вызвано поляризацией диэлектрика, т.е. накоплением отрицательных зарядов вблизи положительно заряженной
- 115. Смещение зарядов под действием поля называется электрической поляризацией вещества Р. Если в единице объема (1см3) содержится
- 116. Основные виды поляризации диэлектрика Молекулярная поляризация P может быть представлена в виде суммы электронной Рэ, атомной
- 117. Электронная поляризация характеризуются упругим смещением электронных орбиталей относительно ядра при воздействии на атом (молекулу) электрического поля
- 118. Атомная поляризация – вид поляризации, когда в электрическом поле смещаются не только электронные облака, но и
- 119. В случае полярных молекул наряду с деформационной поляризуемостью существует еще один вид поляризуемости, вызванной ориентацией постоянных
- 120. Общим выражением для поляризуемости молекул является соотношение: Подставляя значение общей поляризуемости молекул в уравнение Клаузиуса-Моссотти, получим
- 121. Определение дипольных моментов в разбавленных растворах вторым методом Дебая) Основные допущения данного метода: а) В предельно
- 122. Исходя из уравнения Клаузиуса-Моссотти поляризации растворителя и растворенного вещества равны: Следовательно, молярная поляризация раствора равна следующему
- 123. Зная величину Рв-ва∞ можно определить значение ориентационной поляризации растворенного вещества Рэ = R, Pa = 0,1R
- 125. Скачать презентацию

Спектр - в физике — распределение значений физической величины.
Обычно под спектром подразумевается электромагнитный спектр — распределение
Спектр - в физике — распределение значений физической величины.
Обычно под спектром подразумевается электромагнитный спектр — распределение
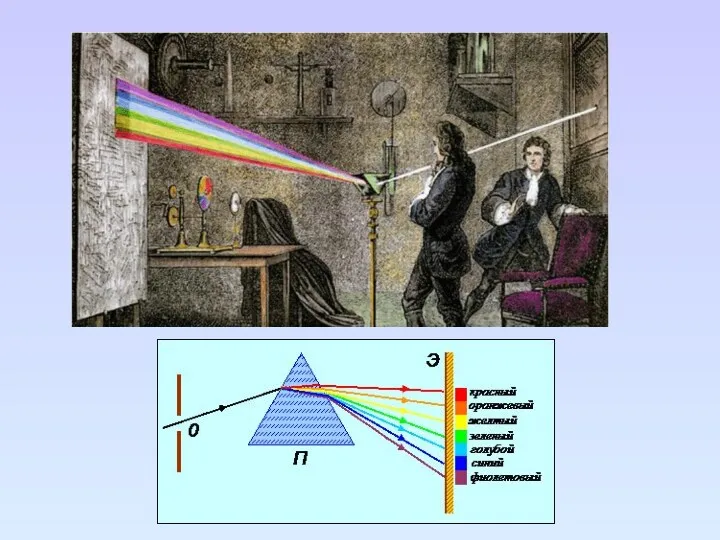

При исследовании строения химических соединений, наибольшую информацию можно получить при
При исследовании строения химических соединений, наибольшую информацию можно получить при

Различные виды излучения характеризуются различной энергией
- энергия излучения, поглощаемая молекулой,
Различные виды излучения характеризуются различной энергией
- энергия излучения, поглощаемая молекулой,

Условия поглощения излучения:
энергия излучения должна совпадать с разностью энергий квантованных
Условия поглощения излучения:
энергия излучения должна совпадать с разностью энергий квантованных

Е*>Е0 происходит поглощение света – спектры поглощения
Е0 > Е* происходит
Е*>Е0 происходит поглощение света – спектры поглощения
Е0 > Е* происходит


Излучение можно охарактеризовать:
длиной волны λ
волновым числом υ
частотой ΰ
Излучение можно охарактеризовать:
длиной волны λ
волновым числом υ
частотой ΰ
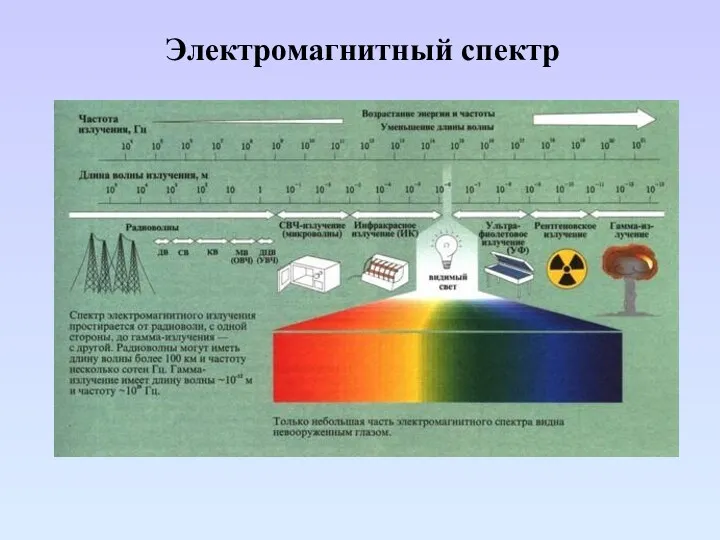
Электромагнитный спектр
Электромагнитный спектр
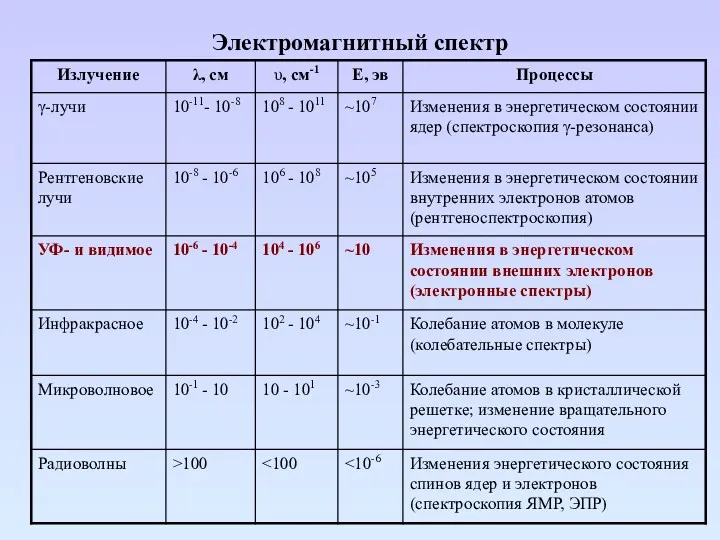
Электромагнитный спектр
Электромагнитный спектр

Чем выше энергия излучения, тем меньше длинна волны и больше частота
Чем выше энергия излучения, тем меньше длинна волны и больше частота

Электронные спектры поглощения возникают в результате переходов электронов из основного в
Электронные спектры поглощения возникают в результате переходов электронов из основного в

Электронные спектры относятся к УФ, видимой и ближней ИК области электромагнитного
Электронные спектры относятся к УФ, видимой и ближней ИК области электромагнитного


Законы поглощения света
Для оптических спектров имеются общие законы поглощения излучения,
Законы поглощения света
Для оптических спектров имеются общие законы поглощения излучения,

k = 0,434k/
- закон Бугера - Ламберта
Первый закон выражает зависимость
k = 0,434k/
- закон Бугера - Ламберта
Первый закон выражает зависимость

Второй закон выражает связь между интенсивностью прошедшего излучения и концентрацией поглощающего
Второй закон выражает связь между интенсивностью прошедшего излучения и концентрацией поглощающего

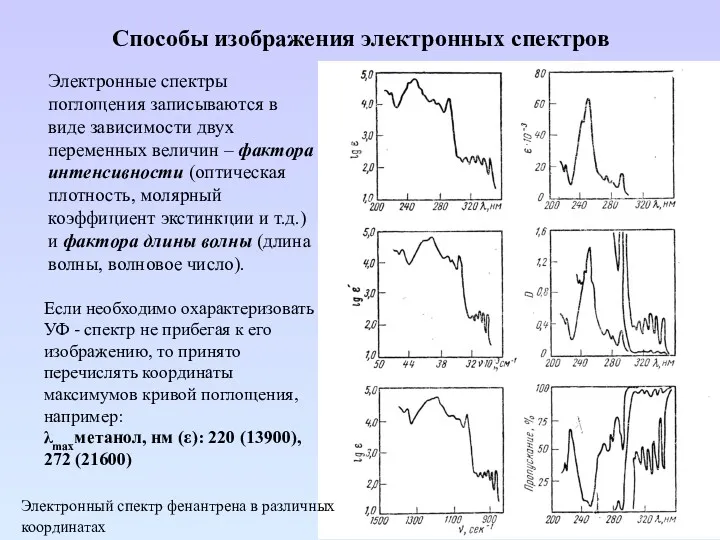
Способы изображения электронных спектров
Электронные спектры поглощения записываются в виде зависимости
Способы изображения электронных спектров
Электронные спектры поглощения записываются в виде зависимости

Природа электронных спектров
При обработке молекулярных спектров пользуются приближением Борна –
Природа электронных спектров
При обработке молекулярных спектров пользуются приближением Борна –
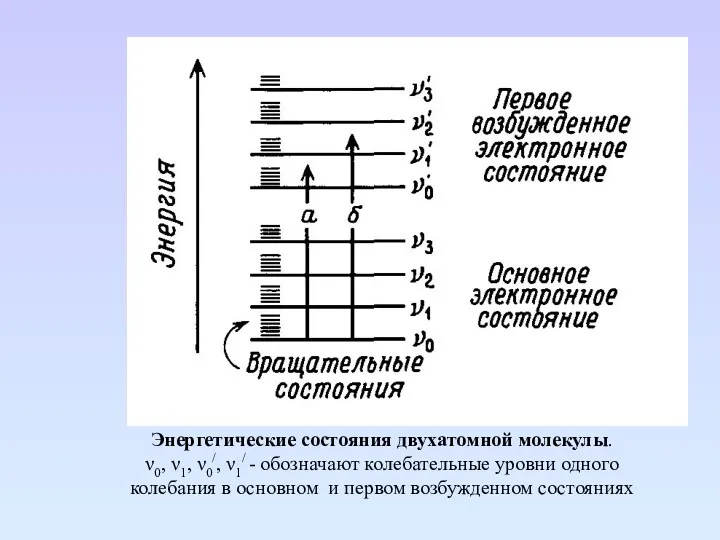
Энергетические состояния двухатомной молекулы.
ν0, ν1, ν0/, ν1/ - обозначают колебательные уровни
Энергетические состояния двухатомной молекулы.
ν0, ν1, ν0/, ν1/ - обозначают колебательные уровни

Колебательные и электронные энергетические уровни двухатомной молекулы
Для каждого электронного состояния
Колебательные и электронные энергетические уровни двухатомной молекулы
Для каждого электронного состояния
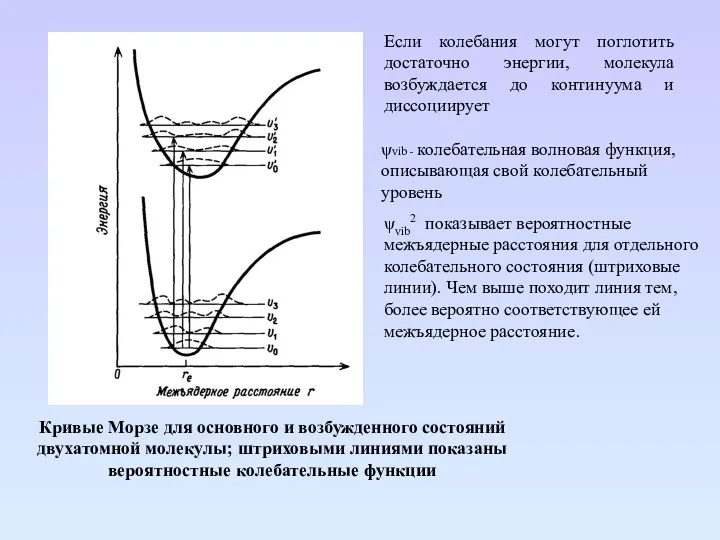
Кривые Морзе для основного и возбужденного состояний двухатомной молекулы; штриховыми линиями
Кривые Морзе для основного и возбужденного состояний двухатомной молекулы; штриховыми линиями

Основные положения электронный спектроскопии
Принцип Франка – Кондона.
За то очень короткое
Основные положения электронный спектроскопии
Принцип Франка – Кондона.
За то очень короткое

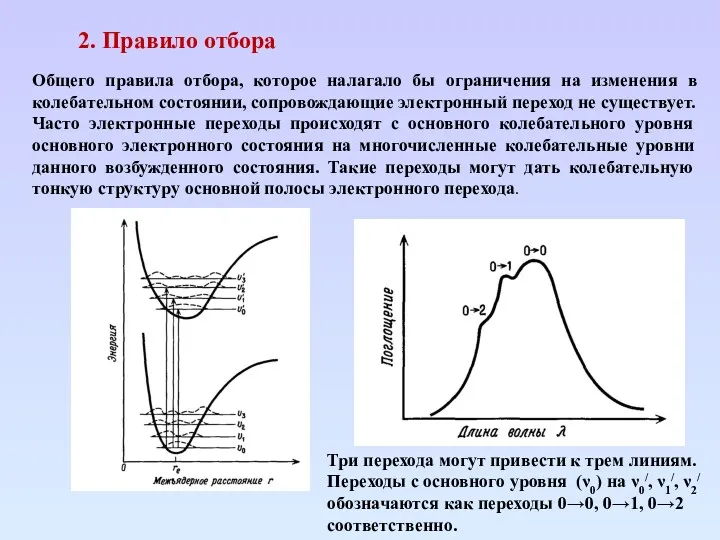
2. Правило отбора
Общего правила отбора, которое налагало бы ограничения на
2. Правило отбора
Общего правила отбора, которое налагало бы ограничения на

Относительная интенсивность различных колебательных подполос зависит от колебательной волновой функции для
Относительная интенсивность различных колебательных подполос зависит от колебательной волновой функции для

3. Требование по симметрии
электронных переходов.
Приведенное выше обсуждение касалось двухатомной
3. Требование по симметрии
электронных переходов.
Приведенное выше обсуждение касалось двухатомной

По аналогии с теорией молекулярных орбиталей:
переход молекулы из основного состояния
По аналогии с теорией молекулярных орбиталей:
переход молекулы из основного состояния

Последовательность энергетических уровней электронов в молекуле органических соединений:
Последовательность энергетических уровней электронов в молекуле органических соединений:

σ → σ* - вакуумная УФ - 100 - 200
σ → σ* - вакуумная УФ - 100 - 200

Элементы симметрии – оси, плоскости, центры инверсии
Операцией симметрии молекулярной
Элементы симметрии – оси, плоскости, центры инверсии
Операцией симметрии молекулярной

Типы симметрии состояний молекулы:
А – симметрия относительно главной поворотной оси
Типы симметрии состояний молекулы:
А – симметрия относительно главной поворотной оси

Электронные переходы в карбонильной группе формальдегида
Относительные энергии молекулярных орбиталей карбонильной группы
Электронные переходы в карбонильной группе формальдегида
Относительные энергии молекулярных орбиталей карбонильной группы
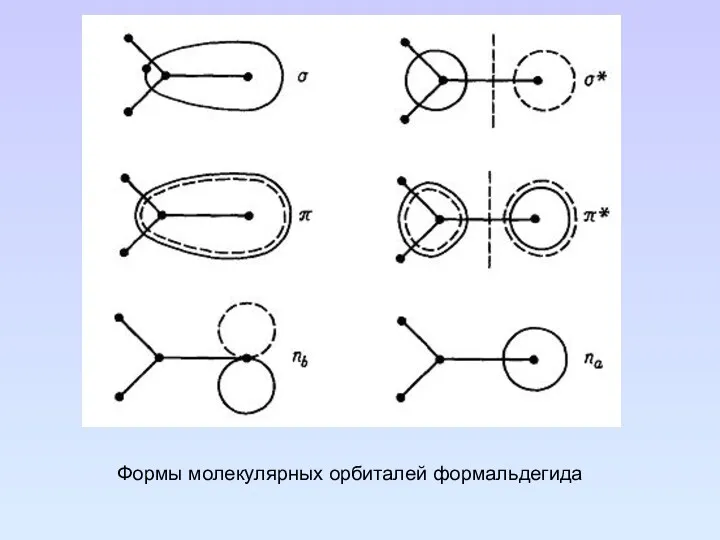
Формы молекулярных орбиталей формальдегида
Формы молекулярных орбиталей формальдегида

Для нахождения симметрии МО определяем полную симметрию молекулы, которая в данном
Для нахождения симметрии МО определяем полную симметрию молекулы, которая в данном

В основном состоянии на каждой МО находится по два электрона и
В основном состоянии на каждой МО находится по два электрона и

Зная симметрию орбиталей в основном состоянии можно определить симметрию возбужденного состояния.
Тип
Зная симметрию орбиталей в основном состоянии можно определить симметрию возбужденного состояния.
Тип


В состоянии, возникающем при переходе n→π*, по одному неспаренному электрону находится
В состоянии, возникающем при переходе n→π*, по одному неспаренному электрону находится

Интенсивность электронных переходов.
Сила осциллятора
υ – волновое число (см-1)
- интегральная
Интенсивность электронных переходов.
Сила осциллятора
υ – волновое число (см-1)
- интегральная

Величина f
безразмерная
для полностью разрешенного перехода f = 1
Интенсивность электронных
Величина f
безразмерная
для полностью разрешенного перехода f = 1
Интенсивность электронных

Сила осциллятора может быть вычислена по формуле:
- вероятность перехода между двумя
Сила осциллятора может быть вычислена по формуле:
- вероятность перехода между двумя

Момент перехода связан с волновыми функциями начального и конечного состояний соотношением:
Момент перехода связан с волновыми функциями начального и конечного состояний соотношением:

не обращается в ноль только в том случае, если подынтегральное
не обращается в ноль только в том случае, если подынтегральное

Разберем на примере электронных переходов в формальдегиде.
1) π→π* переход. Волновая
Разберем на примере электронных переходов в формальдегиде.
1) π→π* переход. Волновая

2) n → π*
А1 → А2. Из таблицы характеров видно,
2) n → π*
А1 → А2. Из таблицы характеров видно,

Правила отбора электронных переходов
1. По мультиплетности. Запрещены переходы между состояниями различной
Правила отбора электронных переходов
1. По мультиплетности. Запрещены переходы между состояниями различной

Перечисленные правила отбора выведены из рассмотрения только электронных волновых функций, без
Перечисленные правила отбора выведены из рассмотрения только электронных волновых функций, без

Хромофоры и ауксохромы
Поглощение вещества в ближнем УФ и видимой области связано
Хромофоры и ауксохромы
Поглощение вещества в ближнем УФ и видимой области связано

1. Очень интенсивные полосы с ε > 103 соответствующие π→π* переходам,
1. Очень интенсивные полосы с ε > 103 соответствующие π→π* переходам,

Замещение или изменение структуры органический соединений вызывает изменение длины волны и
Замещение или изменение структуры органический соединений вызывает изменение длины волны и

Влияние растворителя на электронные спектры веществ
Структура и положение полос поглощения зависят
Влияние растворителя на электронные спектры веществ
Структура и положение полос поглощения зависят
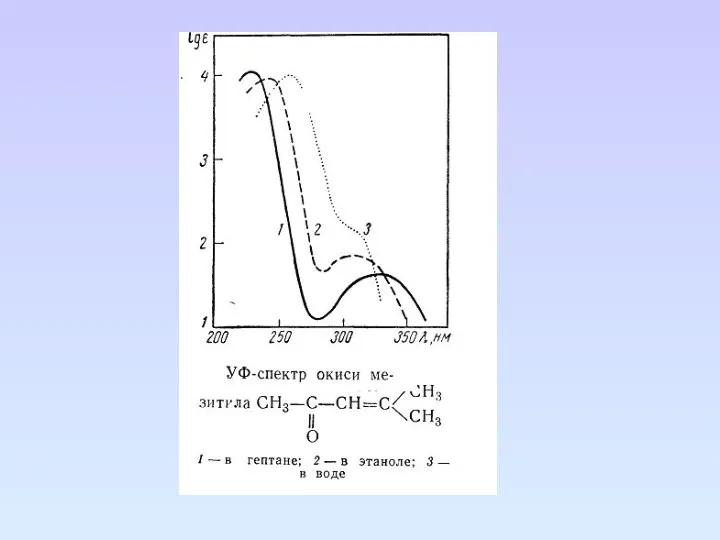
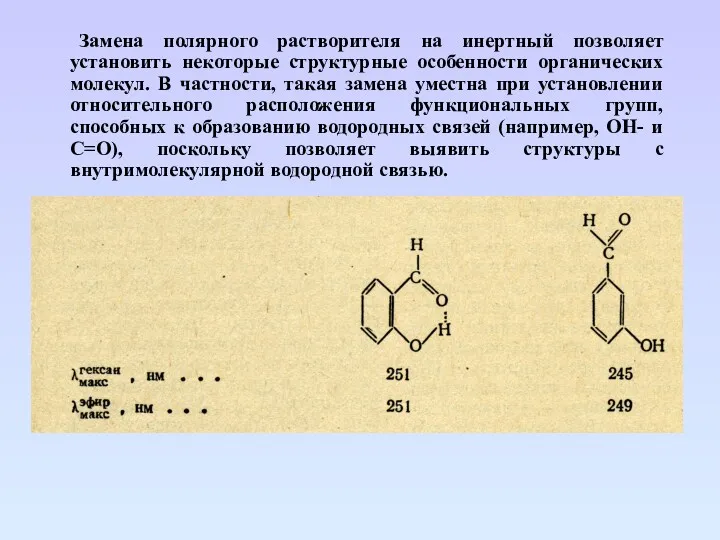
Замена полярного растворителя на инертный позволяет установить некоторые структурные особенности органических
Замена полярного растворителя на инертный позволяет установить некоторые структурные особенности органических
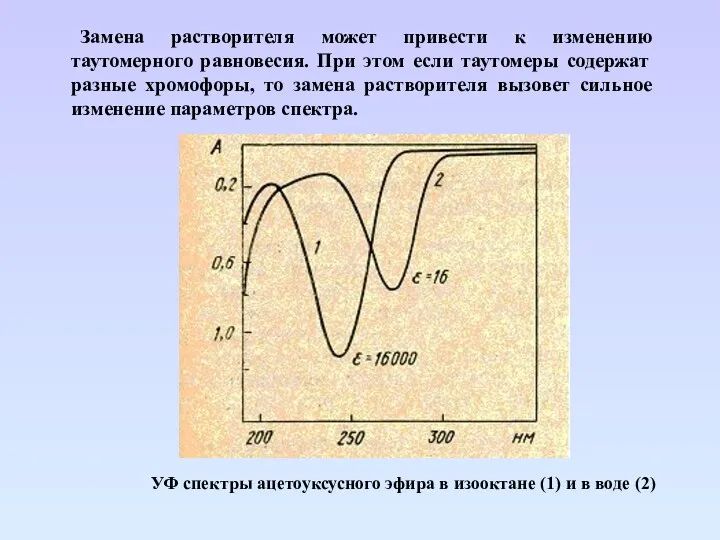
Замена растворителя может привести к изменению таутомерного равновесия. При этом если
Замена растворителя может привести к изменению таутомерного равновесия. При этом если

Использование в качестве растворителе спиртовых и водных растворов кислот и
Использование в качестве растворителе спиртовых и водных растворов кислот и

Для обнаружения хромофоров, содержащих основный азот (например хромофор С=N в алифатических
Для обнаружения хромофоров, содержащих основный азот (например хромофор С=N в алифатических

Электронные спектры поглощения отдельных классов органических соединений
Насыщенные соединения
В насыщенных углеводородах (парафинах
Электронные спектры поглощения отдельных классов органических соединений
Насыщенные соединения
В насыщенных углеводородах (парафинах

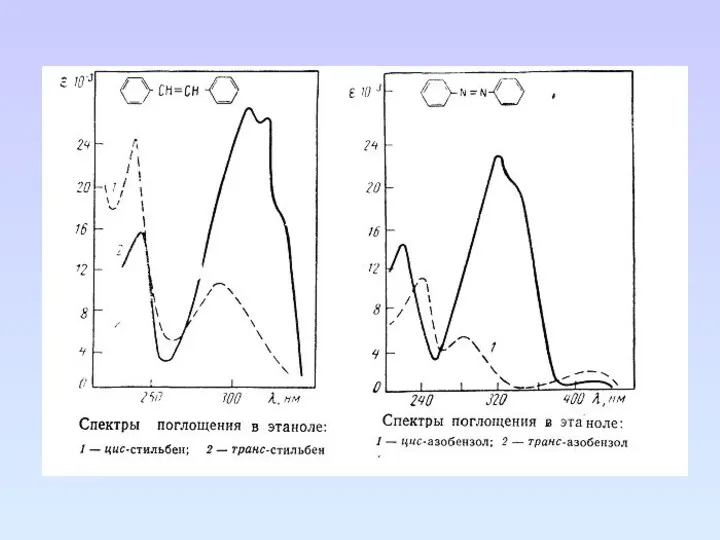
Ненасыщенные соединения
Непредельные углеводороды с изолированными двойными связями имеют интенсивную полосу поглощения,
Ненасыщенные соединения
Непредельные углеводороды с изолированными двойными связями имеют интенсивную полосу поглощения,



Карбонильные соединения
Карбонильные соединения содержат гетероатомы, связанные кратной связью, в таких группах
Карбонильные соединения
Карбонильные соединения содержат гетероатомы, связанные кратной связью, в таких группах
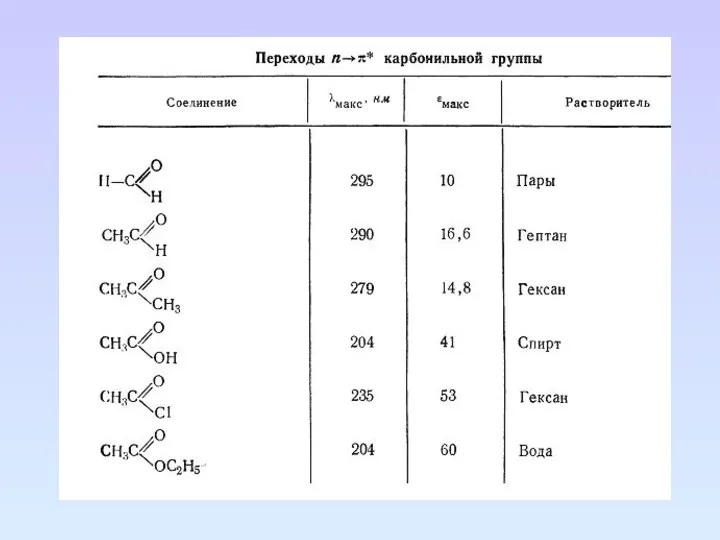
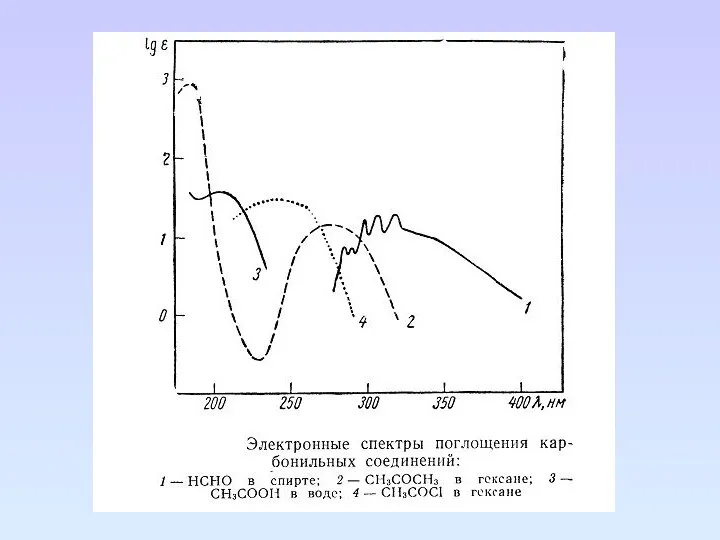

Непредельные карбонильные соединения. Сопряжение кратной связи с карбонильной группой вызывает длинноволновое
Непредельные карбонильные соединения. Сопряжение кратной связи с карбонильной группой вызывает длинноволновое
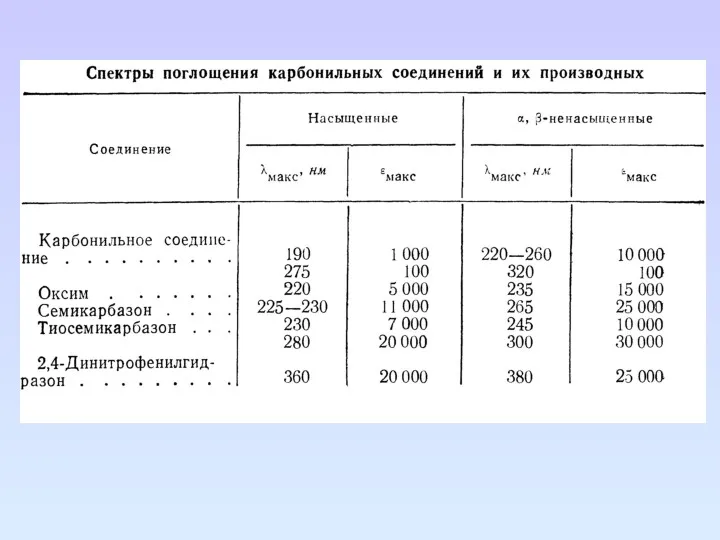

Тиокарбонильные соединения. Тиокарбонильная группа по своему поглощению аналогична карбонильной, однако ее
Тиокарбонильные соединения. Тиокарбонильная группа по своему поглощению аналогична карбонильной, однако ее

Бензол и его производные
Спектр поглощения бензола в гептане
Бензол имеет три полосы
Бензол и его производные
Спектр поглощения бензола в гептане
Бензол имеет три полосы

Алкильные заместители и галогены приводят к небольшому смещению в длинноволновую область
Алкильные заместители и галогены приводят к небольшому смещению в длинноволновую область

При введении в бензольное кольцо полярных групп, содержащих неподеленные электронные пары
При введении в бензольное кольцо полярных групп, содержащих неподеленные электронные пары
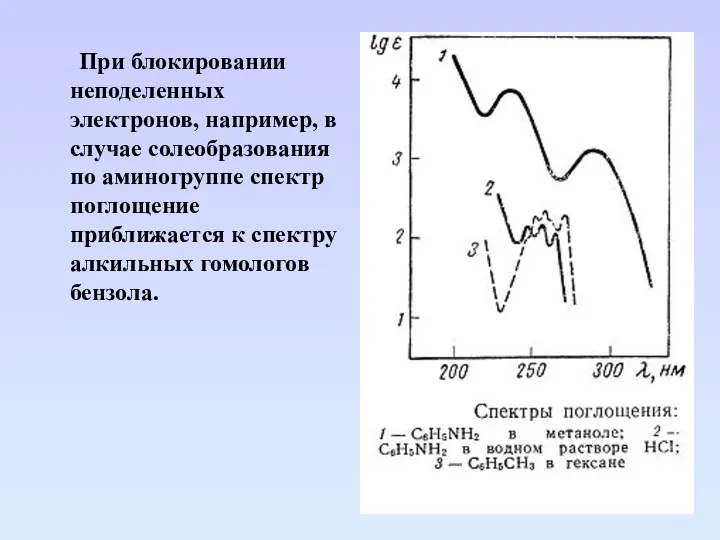
При блокировании неподеленных электронов, например, в случае солеобразования по аминогруппе спектр
При блокировании неподеленных электронов, например, в случае солеобразования по аминогруппе спектр
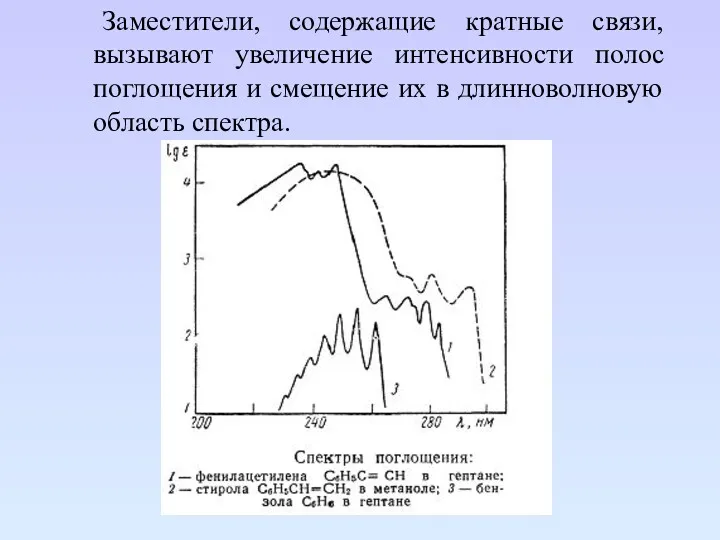
Заместители, содержащие кратные связи, вызывают увеличение интенсивности полос поглощения и смещение
Заместители, содержащие кратные связи, вызывают увеличение интенсивности полос поглощения и смещение

Если заместителем является карбонильная группа, в спектре могут наблюдаться полосы перехода
Если заместителем является карбонильная группа, в спектре могут наблюдаться полосы перехода
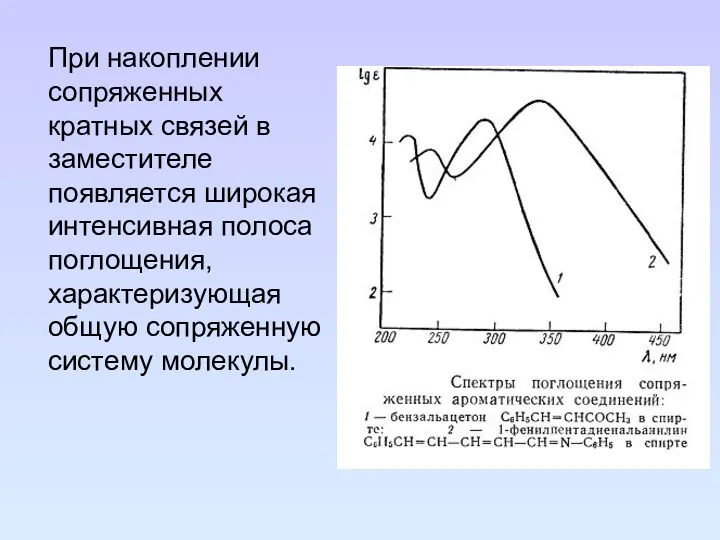
При накоплении сопряженных кратных связей в заместителе появляется широкая интенсивная полоса
При накоплении сопряженных кратных связей в заместителе появляется широкая интенсивная полоса
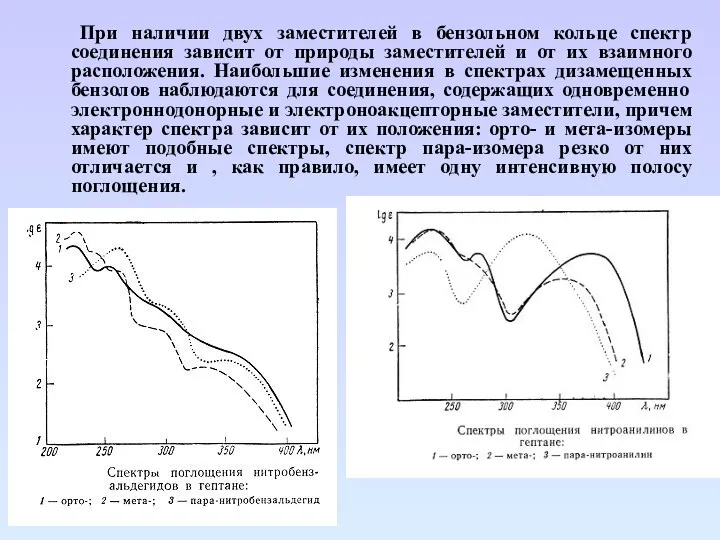
При наличии двух заместителей в бензольном кольце спектр соединения зависит от
При наличии двух заместителей в бензольном кольце спектр соединения зависит от
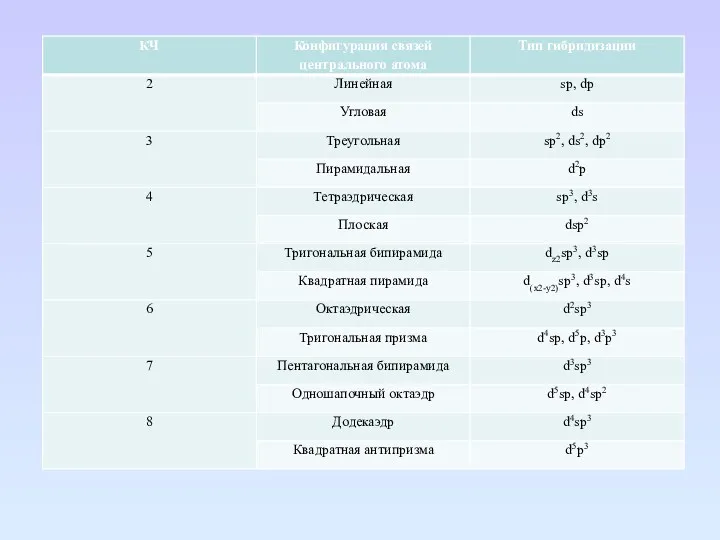

Электронные спектры комплексов переходных металлов
Электронные спектры комплексов переходных металлов интерпретируют с
Электронные спектры комплексов переходных металлов
Электронные спектры комплексов переходных металлов интерпретируют с



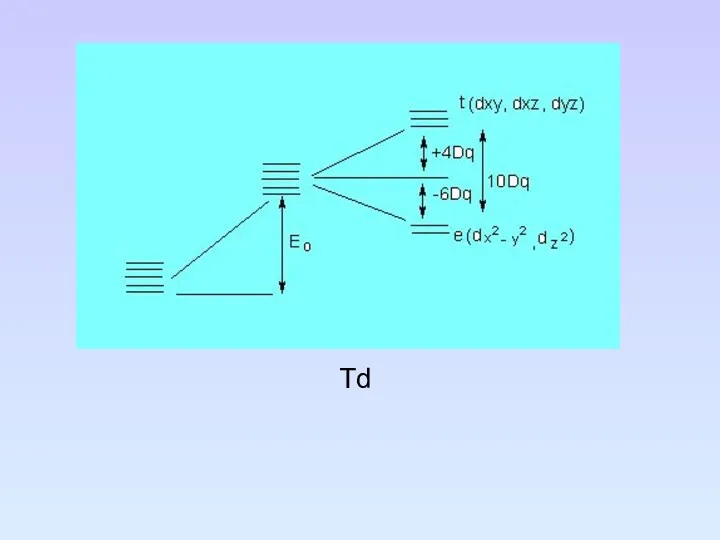
Td
Td
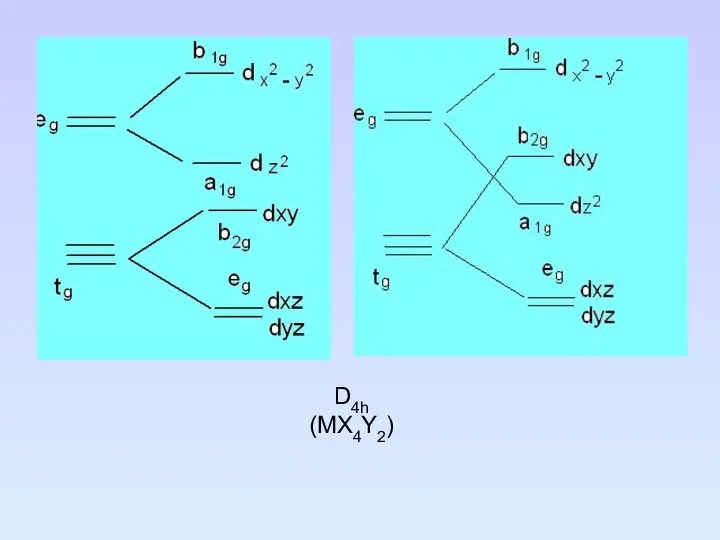
D4h
(MX4Y2)
D4h
(MX4Y2)

В зависимости величины параметра ∆ и межэлектронного взаимодействия зависит распределение электронов
В зависимости величины параметра ∆ и межэлектронного взаимодействия зависит распределение электронов

Порядок величины 10Dq правильно передается энергией самого длинноволнового спектрального перехода
Порядок величины 10Dq правильно передается энергией самого длинноволнового спектрального перехода

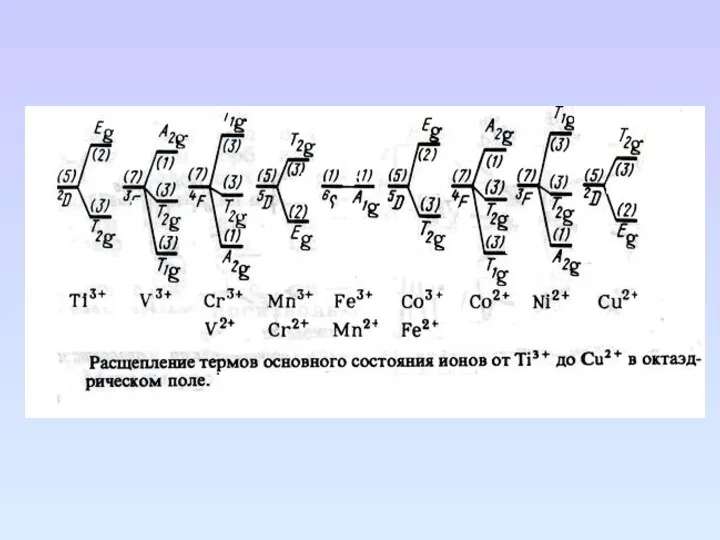
Расщепление энергетических состояний для dn-конфигураций центральных ионов
Определенное энергетическое состояние атома называют
Расщепление энергетических состояний для dn-конфигураций центральных ионов
Определенное энергетическое состояние атома называют

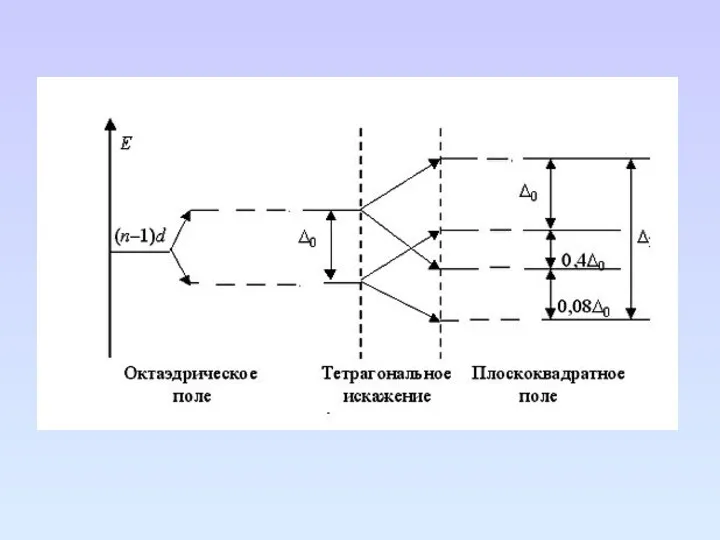
В октаэдрическом и тетраэдрическом, а также кубических электростатических полях лигандов термы
В октаэдрическом и тетраэдрическом, а также кубических электростатических полях лигандов термы
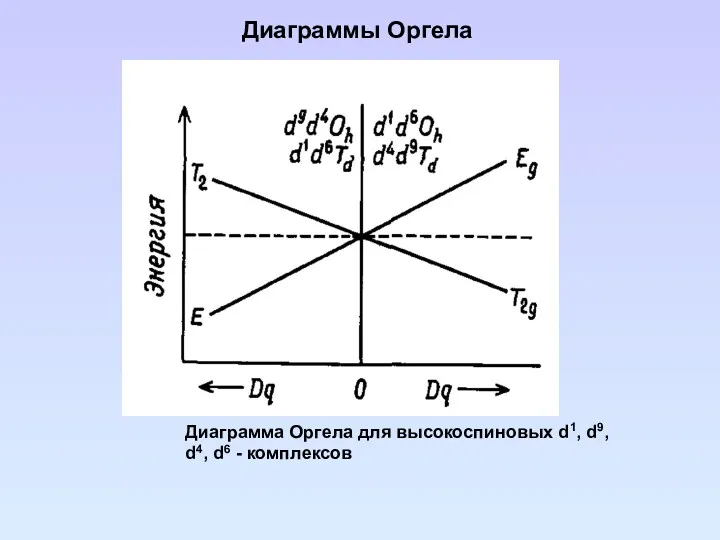
Диаграммы Оргела
Диаграмма Оргела для высокоспиновых d1, d9,
d4, d6 - комплексов
Диаграммы Оргела
Диаграмма Оргела для высокоспиновых d1, d9,
d4, d6 - комплексов
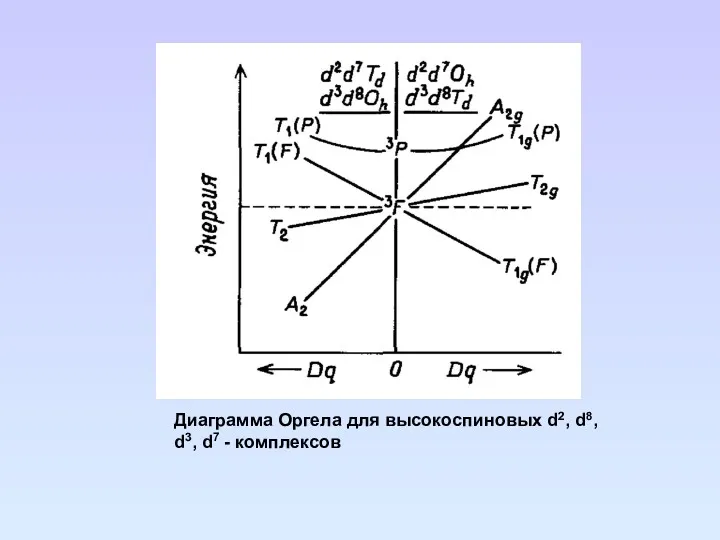
Диаграмма Оргела для высокоспиновых d2, d8,
d3, d7 - комплексов
Диаграмма Оргела для высокоспиновых d2, d8,
d3, d7 - комплексов
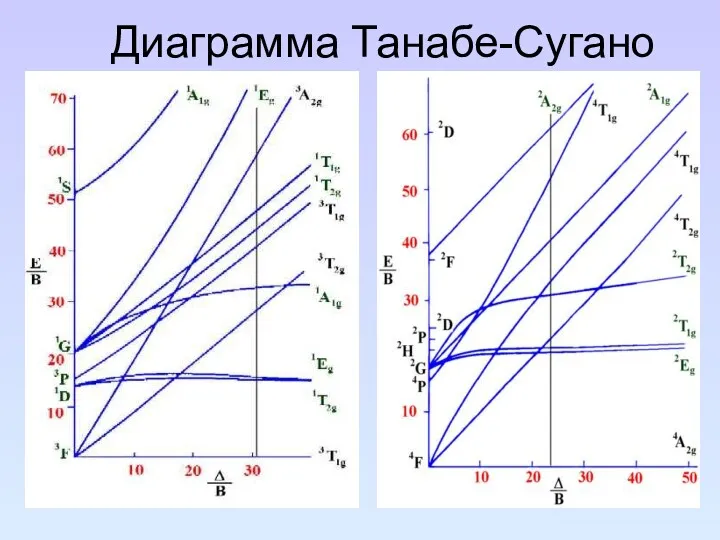
Диаграмма Танабе-Сугано
Диаграмма Танабе-Сугано

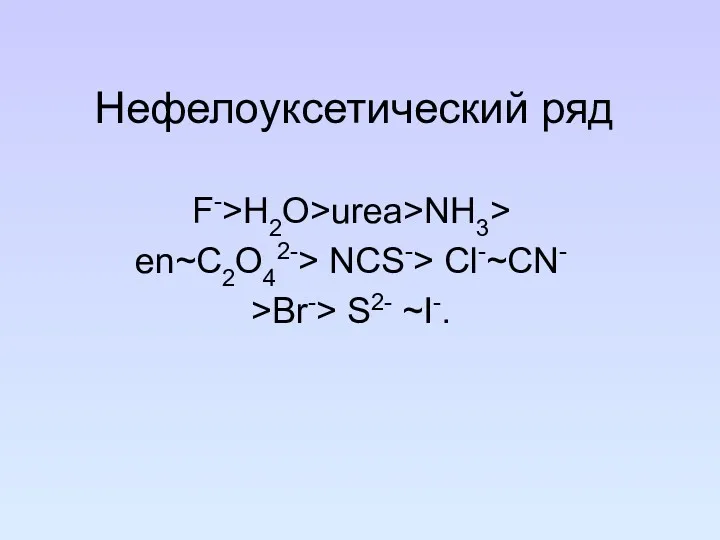
Нефелоуксетический ряд
F->H2O>urea>NH3>
en~C2O42-> NCS-> Cl-~CN-
>Br-> S2- ~I-.
Нефелоуксетический ряд
F->H2O>urea>NH3>
en~C2O42-> NCS-> Cl-~CN-
>Br-> S2- ~I-.

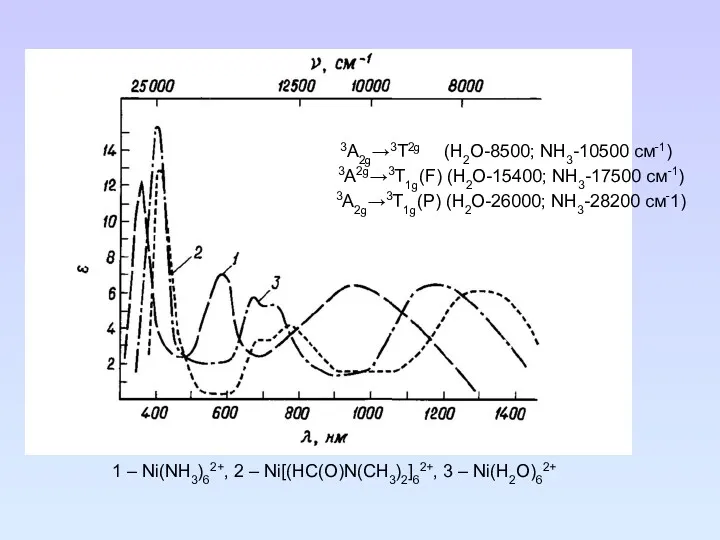
3А2g→3T2g (H2O-8500; NH3-10500 см-1)
3А2g→3T1g(F) (H2O-15400; NH3-17500 см-1)
3А2g→3T1g(P) (H2O-26000; NH3-28200
3А2g→3T2g (H2O-8500; NH3-10500 см-1)
3А2g→3T1g(F) (H2O-15400; NH3-17500 см-1)
3А2g→3T1g(P) (H2O-26000; NH3-28200

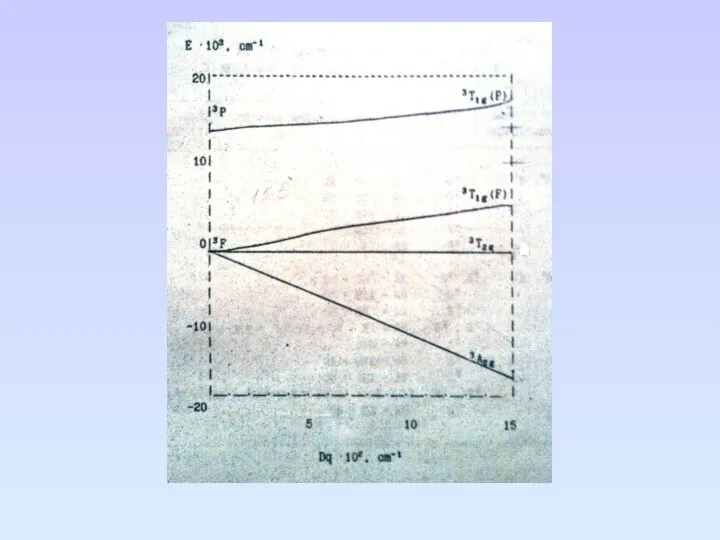
Уравнения Танабе – Сугано для Ni2+ в
октаэдрическом поле
E (3Т2g) =
Уравнения Танабе – Сугано для Ni2+ в
октаэдрическом поле
E (3Т2g) =

Построение диаграммы Оргела
1. Энергию основного состояния центрального иона (энергию терма) принимаем
Построение диаграммы Оргела
1. Энергию основного состояния центрального иона (энергию терма) принимаем

Построение диаграммы Оргела для октаэдрических комплексов Ni2+
Энергию основного состояния Ni2+ т.е
Построение диаграммы Оргела для октаэдрических комплексов Ni2+
Энергию основного состояния Ni2+ т.е

Вычисляем по уравнениям энергии компонент расщепления терма 3F: 3A2g, 3T2g, 3T1g
Вычисляем по уравнениям энергии компонент расщепления терма 3F: 3A2g, 3T2g, 3T1g
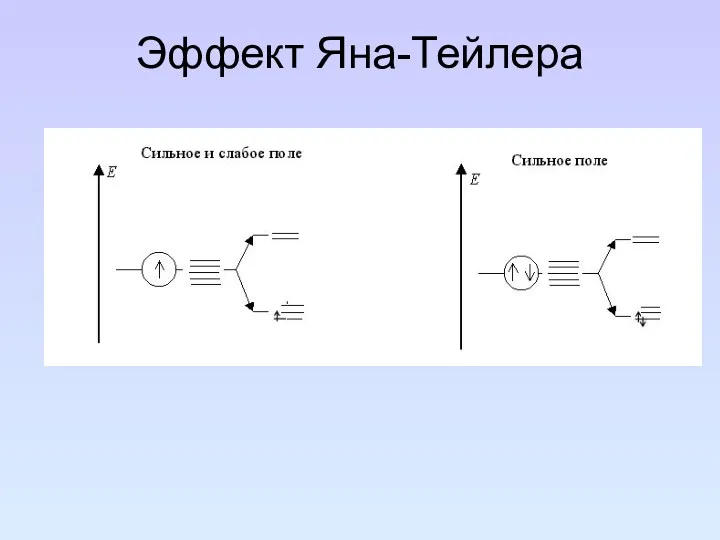
Эффект Яна-Тейлера
Эффект Яна-Тейлера

![[Co(H2O)6]2+ [CoCl4]2+](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/200680/slide-106.jpg)
[Co(H2O)6]2+
[CoCl4]2+
[Co(H2O)6]2+
[CoCl4]2+

Комплексы никеля: 1 – D2d, 2 – Oh, 3 – Td,
Комплексы никеля: 1 – D2d, 2 – Oh, 3 – Td,

Методы определения электрических дипольных моментов молекул
Диэлектрическая проницаемость и электрическая поляризация
Методы определения электрических дипольных моментов молекул
Диэлектрическая проницаемость и электрическая поляризация

Полярным диэлектриком называется такой диэлектрик, молекулы (атомы) которого имеют электроны, расположенные
Полярным диэлектриком называется такой диэлектрик, молекулы (атомы) которого имеют электроны, расположенные
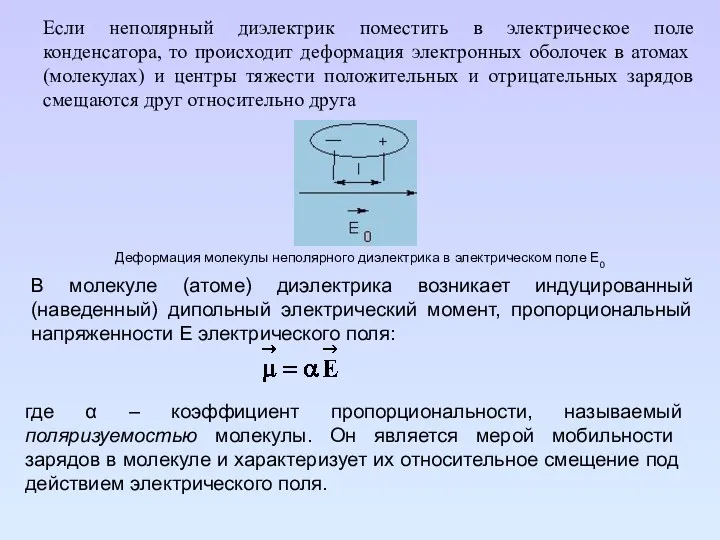
Если неполярный диэлектрик поместить в электрическое поле конденсатора, то происходит деформация
Если неполярный диэлектрик поместить в электрическое поле конденсатора, то происходит деформация

В полярных диэлектриках молекулы представляют собой электрические диполи, которые в отсутствии
В полярных диэлектриках молекулы представляют собой электрические диполи, которые в отсутствии

Заполнение пространства между пластинами конденсатора диэлектриком приводит к уменьшению напряженности поля
Заполнение пространства между пластинами конденсатора диэлектриком приводит к уменьшению напряженности поля

Уменьшение напряженности электрического поля в конденсаторе вызвано поляризацией диэлектрика, т.е. накоплением
Уменьшение напряженности электрического поля в конденсаторе вызвано поляризацией диэлектрика, т.е. накоплением

Смещение зарядов под действием поля называется электрической поляризацией вещества Р. Если
Смещение зарядов под действием поля называется электрической поляризацией вещества Р. Если

Основные виды поляризации диэлектрика
Молекулярная поляризация P может быть представлена в
Основные виды поляризации диэлектрика
Молекулярная поляризация P может быть представлена в

Электронная поляризация характеризуются упругим смещением электронных орбиталей относительно ядра при воздействии
Электронная поляризация характеризуются упругим смещением электронных орбиталей относительно ядра при воздействии

Атомная поляризация – вид поляризации, когда в электрическом поле смещаются не
Атомная поляризация – вид поляризации, когда в электрическом поле смещаются не

В случае полярных молекул наряду с деформационной поляризуемостью существует еще один
В случае полярных молекул наряду с деформационной поляризуемостью существует еще один

Общим выражением для поляризуемости молекул является соотношение:
Подставляя значение общей поляризуемости молекул
Общим выражением для поляризуемости молекул является соотношение:
Подставляя значение общей поляризуемости молекул

Определение дипольных моментов в разбавленных растворах вторым методом Дебая)
Основные допущения данного
Определение дипольных моментов в разбавленных растворах вторым методом Дебая)
Основные допущения данного

Исходя из уравнения Клаузиуса-Моссотти поляризации растворителя и растворенного вещества равны:
Следовательно, молярная
Исходя из уравнения Клаузиуса-Моссотти поляризации растворителя и растворенного вещества равны:
Следовательно, молярная

Зная величину Рв-ва∞ можно определить значение ориентационной поляризации растворенного вещества
Рэ
Зная величину Рв-ва∞ можно определить значение ориентационной поляризации растворенного вещества
Рэ
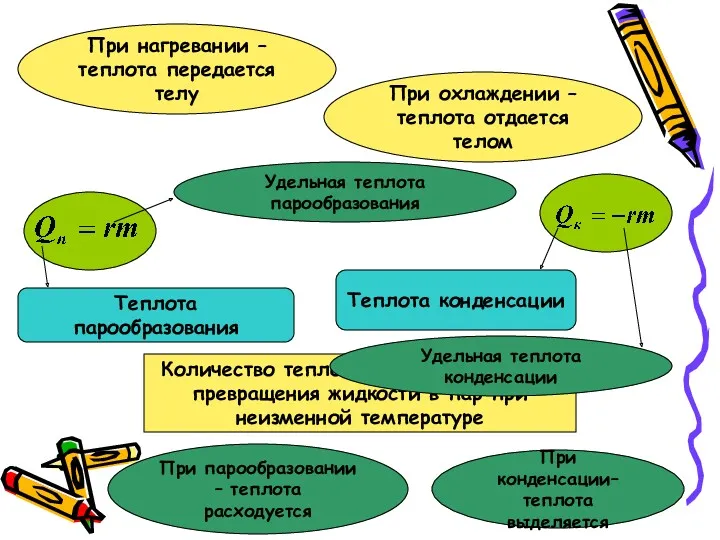 Термодинамика. Теплота
Термодинамика. Теплота Еркін және еріксіз тербелістер. Резонанс
Еркін және еріксіз тербелістер. Резонанс Понятие о технической системе. 6 класс
Понятие о технической системе. 6 класс Урок по теме: Функциональная зависимость
Урок по теме: Функциональная зависимость Medbiophysics as a branch of applied physics. Mechanical vibrations in the medical applications
Medbiophysics as a branch of applied physics. Mechanical vibrations in the medical applications Построение и применение комплексов радиорелейной, тропосферной, спутниковой связи
Построение и применение комплексов радиорелейной, тропосферной, спутниковой связи Силы в природе, законы Ньютона
Силы в природе, законы Ньютона Электростатическое поле в вакууме
Электростатическое поле в вакууме Рулевое управление
Рулевое управление Катушки со сталью в цепи синусоидального тока
Катушки со сталью в цепи синусоидального тока Биологическое действие радиации. Закон радиоактивного распада
Биологическое действие радиации. Закон радиоактивного распада Заттың тығыздығы
Заттың тығыздығы Центр тяжести
Центр тяжести Устойчивость элементов конструкций
Устойчивость элементов конструкций Электрические однофазные цепи синусоидального тока
Электрические однофазные цепи синусоидального тока Курс лекций по сопротивлению материалов (11- 18)
Курс лекций по сопротивлению материалов (11- 18) Электрическое поле в диэлектрике
Электрическое поле в диэлектрике 1. Строение атома
1. Строение атома Газды разряд түрлері
Газды разряд түрлері Литография. Виды литографии
Литография. Виды литографии Эксперимент на уроках физики в основной школе как средство формирования УУД
Эксперимент на уроках физики в основной школе как средство формирования УУД Параметри електричних ланцюгів змінного струму
Параметри електричних ланцюгів змінного струму Законы сохранения в механике
Законы сохранения в механике Электрический ток в различных средах
Электрический ток в различных средах Незаметная бутылка. Эксперимент
Незаметная бутылка. Эксперимент Сдвиг и кручение. Закон Гука при сдвиге. Условие прочности при чистом сдвиге
Сдвиг и кручение. Закон Гука при сдвиге. Условие прочности при чистом сдвиге Кран вспомогательного тормоза №254
Кран вспомогательного тормоза №254 Строительство и эксплуатация зданий и сооружений. Техническая механика
Строительство и эксплуатация зданий и сооружений. Техническая механика