- Основные понятия и определения. Хроматография

Содержание
- 2. 4. Гольдберт К.А., Вигдергауз М.С. Введение в газовую хроматографию. М: Химия, 1990. 352 с. 5. Рудаков
- 3. 9. Пецев Н., Коцев Н. Справочник по газовой хроматографии: Пер. с болг. М:Мир, 1987. 260 с.
- 4. Основные понятия и определения Хроматография – физико-химический метод разделения веществ, основанный на распределении компонентов между двумя
- 5. Основные понятия и определения Сущность метода хроматографии - разделяемые вещества перемещаются через слой неподвижной фазы вместе
- 6. Особенность процесса хроматографирования - многократность повторения сорбции вещества на поверхности сорбента и десорбции с поверхности сорбента,
- 7. Сорбцию можно осуществить двояко: статическая сорбция – процесс протекает при относительном покое обеих фаз и завершается
- 8. Основные понятия и определения Классификация хроматографических методов 1. По агрегатному состоянию фаз
- 9. Основные понятия и определения Классификация хроматографических методов 2. По механизму взаимодействия сорбента и сорбата: - распределительная:
- 10. Основные понятия и определения Классификация хроматографических методов 3. По технике выполнения: - колоночная (набивные и капиллярные
- 11. Основные понятия и определения Классификация хроматографических методов 5. По цели проведения хроматографических процессов: Аналитическая хроматография –
- 12. Хроматограф включает в свой состав: систему подготовки газов (установка, стабилизация и очистка потоков газа); систему подачи
- 13. Разделенные в колонке компоненты с газом-носителем подаются в детектор, который преобразует возникающее изменение физических или физико-химических
- 14. Детектор – прибор непрерывного действия, дающий отклик на соединения в элюате. Комплект современного газового хроматографа содержит
- 15. Интегральный детектор – регистрирует изменение во времени суммарного количества выходящего из колонки компонента, например, общий объем
- 16. Хроматограмма получается в виде ступеней, каждая из которых по высоте пропорциональна количеству компонента, прошедшего через детектор
- 17. Дифференциальный детектор – измеряет мгновенную концентрацию или массовую скорость вещества в потоке газа-носителя. Хроматограмма представляет собой
- 18. При использовании потокового детектора все количество анализируемого компонента успевает однократно зарегистрироваться вне зависимости от скорости пропускания
- 19. Характеристики детекторов Чувствительность – характеризует отношение сигнала детектора к количеству вещества. От чувствительности зависит выбор величины
- 20. Предел детектирования - минимальная концентрация анализируемого вещества в потоке газа-носителя, которая может быть зарегистрирована Сmin. Для
- 21. Нестабильностями нулевой линии являются дрейф и шумы. Дрейф нулевой линии (Base Line Drift). Любое низкочастотное изменение
- 22. Наименьший детектируемый полезный сигнал Минимальным сигналом Еmin, поддающимся измерению, принято считать сигнал, амплитуда которого вдвое превышает
- 23. Концентрация анализируемого вещества, вызывающего этот сигнал, для концентрационного детектора: Сmin= Еmin / Rс = 2 δ
- 24. Для потокового детектора: Сmin= 2 δ / Rj * F , где Rj = A *
- 25. В повседневной практике часто путают понятия «чувствительность» и «предел детектирования», понимая под чувствительностью минимальные концентрации, определяемые
- 26. Одинаковый уровень шума Различный уровень шума Графически это можно выразить:
- 27. Предельные возможности хроматографа в отношении измерения малых концентраций могут быть расширены двумя независимыми путями: увеличением чувствительности
- 28. Линейность – пропорциональность между концентрациями анализируемого вещества на выходе из колонки и сигналом детектора. Диапазон линейности
- 29. Селективность детектора определяют по отношению чувствительности одного детектора к двум веществам S = Rа / Rв
- 30. Воспроизводимость – характеризуется стандартным отклонением серии сигналов при вводе одних и тех же проб. Стабильность работы
- 31. Коэффициент распределения D - описывает равновесие при распределении вещества между неподвижной и подвижной фазами D =
- 32. Теоретические основы хроматографии Основные характеристики
- 33. Характеристики пиков: Время удерживания, ширина и форма Время удерживания tR – время от момента ввода пробы
- 34. tR - не зависит от количества пробы, но зависит от природы вещества и сорбента, а также
- 35. Объем удерживания VR = tR* F, где F – объемная скорость потока Исправленный объем удерживания VR’
- 36. Коэффициент (индекс) удерживания R показывает долю времени нахождения вещества в подвижной фазе R = tm/tR =
- 37. Так как R = Vm/VR , то VR = Vm + DVs D* Vs/Vm называют коэффициентом
- 38. Если k’ значительно меньше 1, то вещество слабо удерживается и продвигается по колонке со скоростью практически
- 39. Исправленный объем удерживания связан с D соотношением VR’ = VR –Vm = D*Vs - основное уравнение
- 40. Коэффициент разделения (селективности) компонентов А и В α = kВ’/kА’ α = DА/DВ α = (tR’)B/(tR’)A
- 41. Разрешение пиков Полнота разделения и правильность определения зависит от того, насколько отделены пики друг от друга,
- 42. Условия раздельной регистрации концентрационного профиля двух компонентов: Достаточное разделение происходит, если ΔtR = 2 (σ1 +
- 43. Для характеристики разделения пиков служит величина, называемая разрешением Rs Rs = (tR(A) –tR(B))*2/(wA +wB) Если wA
- 44. Теоретические основы хроматографии Теория хроматографии Для объяснения явлений, происходящих при хроматографировании, для расчета длины колонок, положения
- 45. Т Мартин и Синг (Нобелевская премия, 1952 г.) ввели понятие высоты, эквивалентной теоретической тарелке H, (ВЭТТ)
- 46. Высота тарелки и число теоретических тарелок характеризуют эффективность колонки. Высота, эквивалентная теоретической тарелке, рассчитывается: Дисперсия σL2
- 47. Теоретические основы хроматографии Теория хроматографии Для ширины у основания w = 4 σt , с учетом
- 48. Если коэффициенты емкости близки, то часто используется уравнение в упрощенной форме: Если известно разрешение, то зная
- 49. Однако теория теоретических тарелок не позволяет выявить зависимости N и Н от скорости подачи подвижной фазы,
- 50. Хроматограмма отражает статистическое поведение множества молекул, а не индивидуальной молекулы. Из-за случайного стечения обстоятельств одни молекулы
- 51. Теоретический подход, объясняющий размывание пиков, основан на изучении форм изотерм сорбции – графической зависимости количества вещества
- 52. Теоретические основы хроматографии Теория хроматографии Выпуклая изотерма свидетельствует о том, что значение D для больших концентраций
- 53. Обычно работают в областях, характеризующихся линейной изотермой. На продвижение частиц влияет ряд факторов, искажающих форму пика
- 54. Влияние этих факторов на эффективность колонки учитывает кинетическая теория, разработанная Ван-Деемтером. Согласно этой теории размывание пиков
- 55. Вихревая диффузия связана со структурой сорбента и изменяется по длине колонки. А – описывает расстояние ,
- 56. Молекулярная (продольная) диффузия обусловливает размывание полос из-за миграции молекул в подвижной фазе из участков с большей
- 57. Dm в жидкости значительно ниже, чем в газе, поэтому массообмен в жидкостной хроматографии и В не
- 58. Диффузия уменьшается при увеличении линейной скорости подвижной фазы, Н – уменьшается при увеличении линейной скорости из-за
- 59. Сопротивление массопереносу Сv учитывает размывание пика за счет сопротивления массопереносу при непрерывном переходе вещества из подвижной
- 60. Теоретические основы хроматографии Теория хроматографии Приравнивая к нулю dH/dv, получаем скорость, отвечающую минимальному значению Н: Откуда:
- 61. Эффективное разделение за более короткое время достигается при небольшой высоте тарелки Н. Нmin – достигается: Малым
- 62. В ГХ коэффициенты диффузии в подвижной фазе могут быть значительно уменьшены снижением температуры. Поскольку коэффициенты диффузии
- 63. Количественный анализ основан на зависимости между площадью S (или высотой h) пика и количеством определяемого компонента
- 64. [S]=[мм2];[A*с];[В*с] [h]=[мм] Анализ и методы расчета хроматограмм
- 65. Наиболее просто измеряются и рассчитываются параметры гауссовых пиков. Контур этих пиков описывается уравнением: Анализ и методы
- 66. Анализ и методы расчета хроматограмм
- 67. Расчет площади пика как площади, ограниченной гауссовой кривой (для симметричных пиков), проводят по формуле, полученной интегрированием
- 68. Ассиметричность пика может быть вызвана: 1. Перегрузкой колонки анализируемым веществом (рис. а); 2. Наличием остаточной адсорбционной
- 69. Для численного выражения ассиметричности используют коэффициент ассиметричности: Анализ и методы расчета хроматограмм Для симметричного пика: Kas=1
- 70. Методы триангуляции Измерение площадей гауссовых пиков а). S=1/2·h’·b0 Метод дает 96,8% от площади гауссова пика. Недостатки:
- 71. Анализ и методы расчета хроматограмм б). S=1/2·h·b0 Метод дает 80% от площади гауссова пика. в). S=h·b0,5
- 72. Истинная площадь гауссова пика может быть найдена: Sист=h·b0.368 Анализ и методы расчета хроматограмм
- 73. Количественный состав пробы определяют: Методом нормировки (внутренней нормализации) Методом внешней стандартизации (абсолютной градуировки) Методом внутренней стандартизации
- 74. Метод нормировки применяется наиболее часто. Для его применения необходимо, чтобы на хроматограмме были зарегистрированы все компоненты,
- 75. При анализе смеси трех компонентов, относительное содержание компонентов рассчитывают Х,% = 100 % * Sx /(Sx
- 76. Если чувствительность детектора различна по отношению к каждому компоненту пробы, то используют поправочные коэффициенты f: х,%
- 77. Метод внешней стандартизации (абсолютной градуировки) Используют при определении отдельных веществ в простых смесях, при определении микропримесей
- 78. Метод внутренней стандартизации применяют при отсутствии на хроматограмме пиков некоторых компонентов смеси Метод основан на том,
- 79. Сi =k*Сст* Si /Sст; x% =100 %*k*r* Si /Sст Поправочный коэффициент k рассчитывают по стандартной смеси
- 80. Качественный анализ Время удерживания, объем удерживания - характеризуют природу анализируемого вещества Идентификация по времени удерживания Совпадение
- 81. Идентификация по относительному времени удерживания Часто для идентификации используют величину относительного удерживания, зависящую только от состава
- 82. Идентификация по индексам удерживания Ковача За стандарт берут два соседних алкана, один из которых элюируется до,
- 83. Идентификация по линейным зависимостям параметров удерживания в гомологическом ряду органических соединений: lgVR’= А + Bz lgVR’=A
- 85. Скачать презентацию

4. Гольдберт К.А., Вигдергауз М.С. Введение в газовую хроматографию. М: Химия,

9. Пецев Н., Коцев Н. Справочник по газовой хроматографии: Пер. с
9. Пецев Н., Коцев Н. Справочник по газовой хроматографии: Пер. с

Основные понятия и определения
Хроматография
– физико-химический метод разделения веществ, основанный на распределении
Основные понятия и определения
Хроматография
– физико-химический метод разделения веществ, основанный на распределении

Основные понятия и определения
Сущность метода хроматографии - разделяемые вещества перемещаются через
Основные понятия и определения
Сущность метода хроматографии - разделяемые вещества перемещаются через

Особенность процесса хроматографирования - многократность повторения сорбции вещества на поверхности сорбента

Сорбцию можно осуществить двояко:
статическая сорбция – процесс протекает при относительном
Сорбцию можно осуществить двояко:
статическая сорбция – процесс протекает при относительном

Основные понятия и определения
Классификация хроматографических методов
1. По агрегатному состоянию фаз
Основные понятия и определения
Классификация хроматографических методов
1. По агрегатному состоянию фаз

Основные понятия и определения
Классификация хроматографических методов
2. По механизму взаимодействия сорбента и
Основные понятия и определения
Классификация хроматографических методов
2. По механизму взаимодействия сорбента и

Основные понятия и определения
Классификация хроматографических методов
3. По технике выполнения:
- колоночная (набивные
Основные понятия и определения
Классификация хроматографических методов
3. По технике выполнения:
- колоночная (набивные

Основные понятия и определения
Классификация хроматографических методов
5. По цели проведения хроматографических
Основные понятия и определения
Классификация хроматографических методов
5. По цели проведения хроматографических

Хроматограф включает в свой состав:
систему подготовки газов (установка, стабилизация и очистка
Хроматограф включает в свой состав:
систему подготовки газов (установка, стабилизация и очистка

Разделенные в колонке компоненты с газом-носителем подаются в детектор, который преобразует
Разделенные в колонке компоненты с газом-носителем подаются в детектор, который преобразует

Детектор – прибор непрерывного действия, дающий отклик на соединения в элюате.
Детектор – прибор непрерывного действия, дающий отклик на соединения в элюате.

Интегральный детектор – регистрирует изменение во времени суммарного количества выходящего из
Интегральный детектор – регистрирует изменение во времени суммарного количества выходящего из
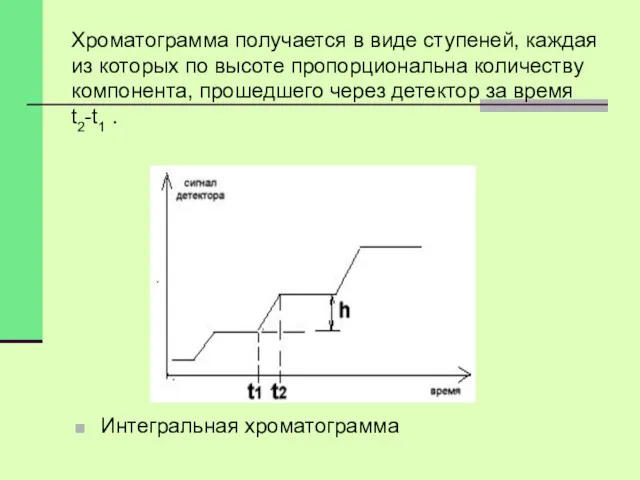
Хроматограмма получается в виде ступеней, каждая из которых по высоте пропорциональна
Хроматограмма получается в виде ступеней, каждая из которых по высоте пропорциональна
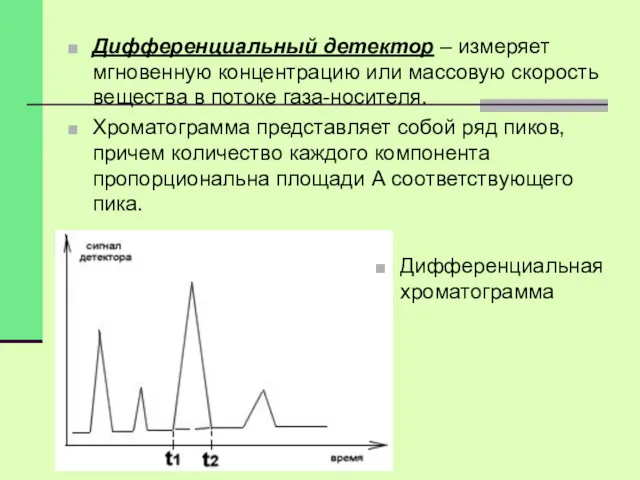
Дифференциальный детектор – измеряет мгновенную концентрацию или массовую скорость вещества в
Дифференциальный детектор – измеряет мгновенную концентрацию или массовую скорость вещества в

При использовании потокового детектора все количество анализируемого компонента успевает однократно зарегистрироваться
При использовании потокового детектора все количество анализируемого компонента успевает однократно зарегистрироваться

Характеристики детекторов
Чувствительность – характеризует отношение сигнала детектора к количеству вещества.
От чувствительности
Характеристики детекторов
Чувствительность – характеризует отношение сигнала детектора к количеству вещества.
От чувствительности

Предел детектирования - минимальная концентрация анализируемого вещества в потоке газа-носителя, которая
Предел детектирования - минимальная концентрация анализируемого вещества в потоке газа-носителя, которая

Нестабильностями нулевой линии являются дрейф и шумы.
Дрейф нулевой линии (Base Line
Нестабильностями нулевой линии являются дрейф и шумы.
Дрейф нулевой линии (Base Line

Наименьший детектируемый полезный сигнал
Минимальным сигналом Еmin, поддающимся измерению, принято считать сигнал,
Наименьший детектируемый полезный сигнал
Минимальным сигналом Еmin, поддающимся измерению, принято считать сигнал,

Концентрация анализируемого вещества, вызывающего этот сигнал, для концентрационного детектора:
Сmin= Еmin /
Концентрация анализируемого вещества, вызывающего этот сигнал, для концентрационного детектора:
Сmin= Еmin /

Для потокового детектора:
Сmin= 2 δ / Rj * F , где
Rj
Для потокового детектора:
Сmin= 2 δ / Rj * F , где
Rj

В повседневной практике часто путают понятия «чувствительность» и «предел детектирования», понимая
В повседневной практике часто путают понятия «чувствительность» и «предел детектирования», понимая
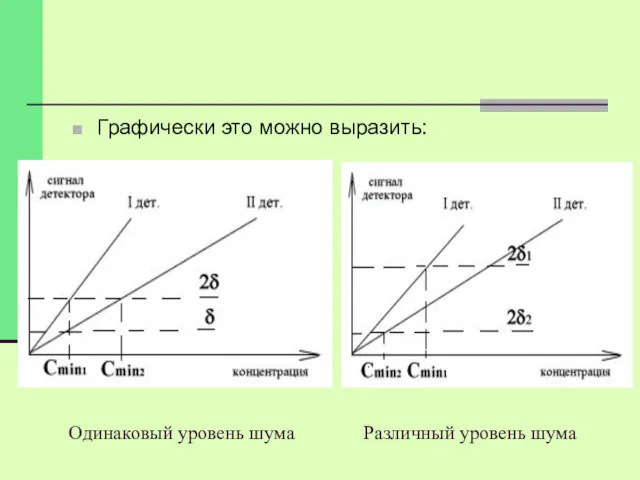
Одинаковый уровень шума Различный уровень шума
Графически это можно выразить:
Одинаковый уровень шума Различный уровень шума
Графически это можно выразить:

Предельные возможности хроматографа в отношении измерения малых концентраций могут быть расширены
Предельные возможности хроматографа в отношении измерения малых концентраций могут быть расширены
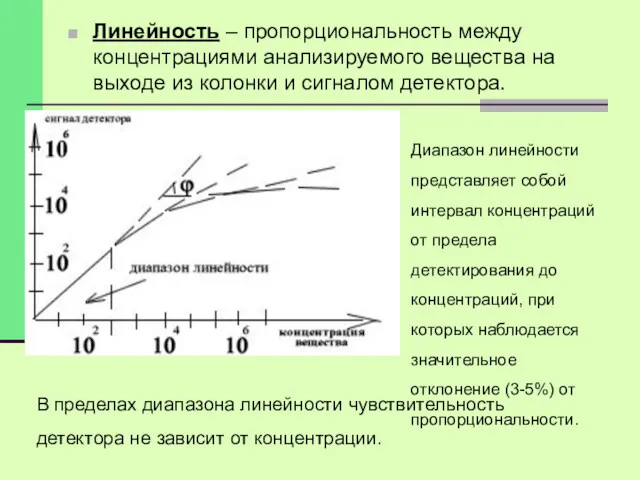
Линейность – пропорциональность между концентрациями анализируемого вещества на выходе из колонки
Линейность – пропорциональность между концентрациями анализируемого вещества на выходе из колонки

Селективность детектора
определяют по отношению чувствительности одного детектора к двум веществам
S
Селективность детектора
определяют по отношению чувствительности одного детектора к двум веществам
S

Воспроизводимость – характеризуется стандартным отклонением серии сигналов при вводе одних и
Воспроизводимость – характеризуется стандартным отклонением серии сигналов при вводе одних и

Коэффициент распределения D - описывает равновесие при распределении вещества между неподвижной
Коэффициент распределения D - описывает равновесие при распределении вещества между неподвижной
Теоретические основы хроматографии
Основные характеристики
Теоретические основы хроматографии
Основные характеристики

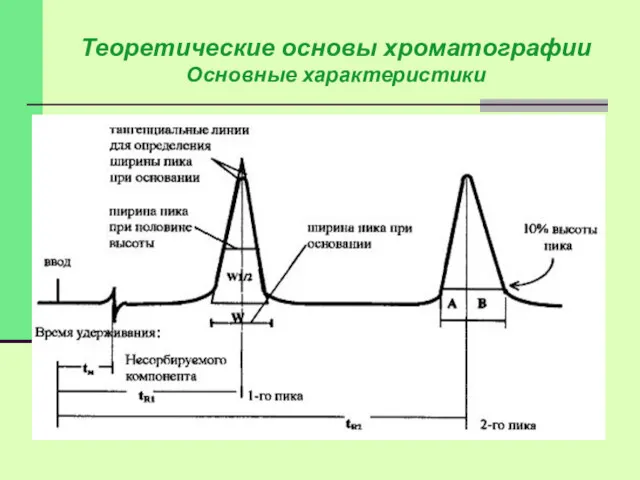
Характеристики пиков:
Время удерживания, ширина и форма
Время удерживания tR – время
Характеристики пиков:
Время удерживания, ширина и форма
Время удерживания tR – время

tR - не зависит от количества пробы, но зависит от природы
tR - не зависит от количества пробы, но зависит от природы

Объем удерживания VR = tR* F,
где F – объемная скорость потока
Исправленный
Объем удерживания VR = tR* F,
где F – объемная скорость потока
Исправленный

Коэффициент (индекс) удерживания R показывает долю времени нахождения вещества в подвижной
Коэффициент (индекс) удерживания R показывает долю времени нахождения вещества в подвижной

Так как R = Vm/VR , то
VR = Vm
Так как R = Vm/VR , то
VR = Vm

Если k’ значительно меньше 1, то вещество слабо удерживается и продвигается
Если k’ значительно меньше 1, то вещество слабо удерживается и продвигается

Исправленный объем удерживания связан с D соотношением
VR’ = VR –Vm
Исправленный объем удерживания связан с D соотношением
VR’ = VR –Vm

Коэффициент разделения (селективности) компонентов А и В
α = kВ’/kА’
Коэффициент разделения (селективности) компонентов А и В
α = kВ’/kА’
Разрешение пиков
Полнота разделения и правильность определения зависит от того, насколько отделены
Разрешение пиков
Полнота разделения и правильность определения зависит от того, насколько отделены

Условия раздельной регистрации концентрационного профиля двух компонентов:
Достаточное разделение происходит, если
ΔtR
Условия раздельной регистрации концентрационного профиля двух компонентов:
Достаточное разделение происходит, если
ΔtR
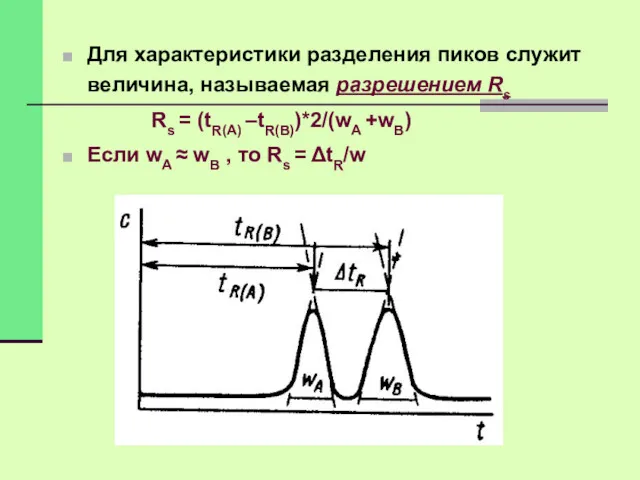
Для характеристики разделения пиков служит величина, называемая разрешением Rs
Rs =
Для характеристики разделения пиков служит величина, называемая разрешением Rs
Rs =

Теоретические основы хроматографии
Теория хроматографии
Для объяснения явлений, происходящих при хроматографировании, для расчета
Теоретические основы хроматографии
Теория хроматографии
Для объяснения явлений, происходящих при хроматографировании, для расчета

Т
Мартин и Синг (Нобелевская премия, 1952 г.) ввели понятие высоты, эквивалентной
Т
Мартин и Синг (Нобелевская премия, 1952 г.) ввели понятие высоты, эквивалентной

Высота тарелки и число теоретических тарелок характеризуют эффективность колонки.
Высота,
Высота тарелки и число теоретических тарелок характеризуют эффективность колонки.
Высота,

Теоретические основы хроматографии
Теория хроматографии
Для ширины у основания w = 4 σt
Теоретические основы хроматографии
Теория хроматографии
Для ширины у основания w = 4 σt

Если коэффициенты емкости близки, то часто используется уравнение в упрощенной форме:
Если
Если коэффициенты емкости близки, то часто используется уравнение в упрощенной форме:
Если

Однако теория теоретических тарелок не позволяет выявить зависимости N и Н
Однако теория теоретических тарелок не позволяет выявить зависимости N и Н

Хроматограмма отражает статистическое поведение множества молекул, а не индивидуальной молекулы.
Из-за случайного
Хроматограмма отражает статистическое поведение множества молекул, а не индивидуальной молекулы.
Из-за случайного

Теоретический подход, объясняющий размывание пиков, основан на изучении форм изотерм сорбции
Теоретический подход, объясняющий размывание пиков, основан на изучении форм изотерм сорбции
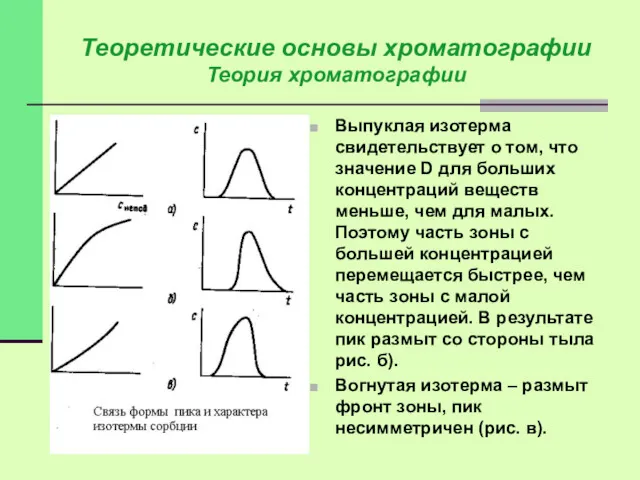
Теоретические основы хроматографии
Теория хроматографии
Выпуклая изотерма свидетельствует о том, что значение D
Теоретические основы хроматографии
Теория хроматографии
Выпуклая изотерма свидетельствует о том, что значение D

Обычно работают в областях, характеризующихся линейной изотермой.
На продвижение частиц влияет ряд
Обычно работают в областях, характеризующихся линейной изотермой.
На продвижение частиц влияет ряд

Влияние этих факторов на эффективность колонки учитывает кинетическая теория, разработанная Ван-Деемтером.
Согласно
Влияние этих факторов на эффективность колонки учитывает кинетическая теория, разработанная Ван-Деемтером.
Согласно

Вихревая диффузия связана со структурой сорбента и изменяется по длине колонки.
Вихревая диффузия связана со структурой сорбента и изменяется по длине колонки.

Молекулярная (продольная) диффузия
обусловливает размывание полос из-за миграции молекул в
Молекулярная (продольная) диффузия
обусловливает размывание полос из-за миграции молекул в

Dm в жидкости значительно ниже, чем в газе, поэтому массообмен в
Dm в жидкости значительно ниже, чем в газе, поэтому массообмен в

Диффузия уменьшается при увеличении линейной скорости подвижной фазы,
Н – уменьшается при
Диффузия уменьшается при увеличении линейной скорости подвижной фазы,
Н – уменьшается при

Сопротивление массопереносу Сv учитывает размывание пика за счет сопротивления массопереносу при
Сопротивление массопереносу Сv учитывает размывание пика за счет сопротивления массопереносу при
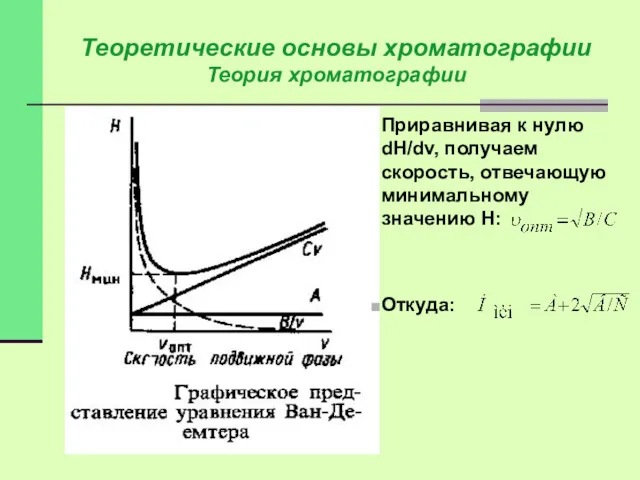
Теоретические основы хроматографии
Теория хроматографии
Приравнивая к нулю dH/dv, получаем скорость, отвечающую минимальному
Теоретические основы хроматографии
Теория хроматографии
Приравнивая к нулю dH/dv, получаем скорость, отвечающую минимальному

Эффективное разделение за более короткое время достигается при небольшой высоте тарелки
Эффективное разделение за более короткое время достигается при небольшой высоте тарелки

В ГХ коэффициенты диффузии в подвижной фазе могут быть значительно уменьшены
В ГХ коэффициенты диффузии в подвижной фазе могут быть значительно уменьшены
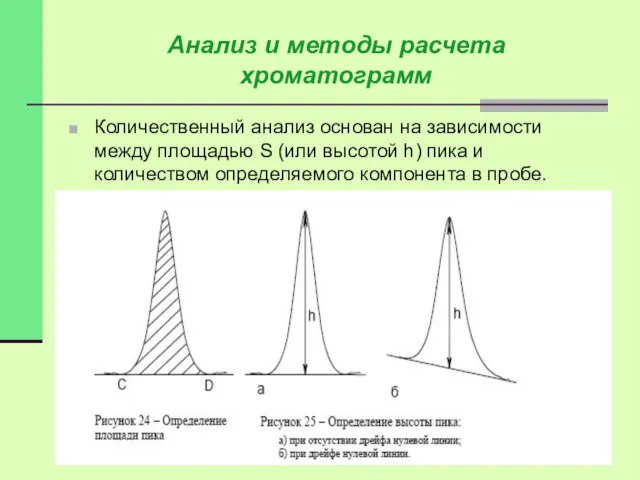
Количественный анализ основан на зависимости между площадью S (или высотой h)
Количественный анализ основан на зависимости между площадью S (или высотой h)
![[S]=[мм2];[A*с];[В*с] [h]=[мм] Анализ и методы расчета хроматограмм](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/144821/slide-63.jpg)
[S]=[мм2];[A*с];[В*с]
[h]=[мм]
Анализ и методы расчета хроматограмм
[S]=[мм2];[A*с];[В*с]
[h]=[мм]
Анализ и методы расчета хроматограмм

Наиболее просто измеряются и рассчитываются параметры гауссовых пиков.
Контур этих пиков описывается
Наиболее просто измеряются и рассчитываются параметры гауссовых пиков.
Контур этих пиков описывается
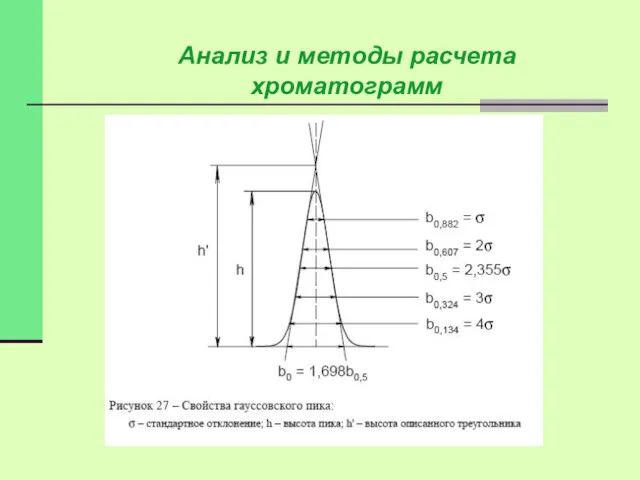
Анализ и методы расчета хроматограмм
Анализ и методы расчета хроматограмм

Расчет площади пика как площади, ограниченной гауссовой кривой (для симметричных пиков),
Расчет площади пика как площади, ограниченной гауссовой кривой (для симметричных пиков),
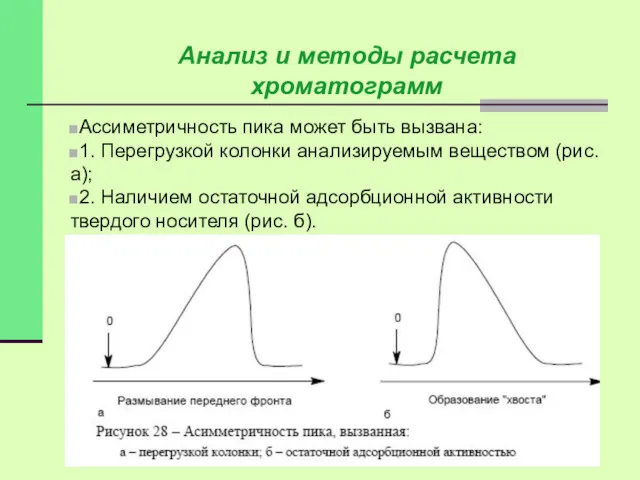
Ассиметричность пика может быть вызвана:
1. Перегрузкой колонки анализируемым веществом (рис. а);
2.
Ассиметричность пика может быть вызвана:
1. Перегрузкой колонки анализируемым веществом (рис. а);
2.
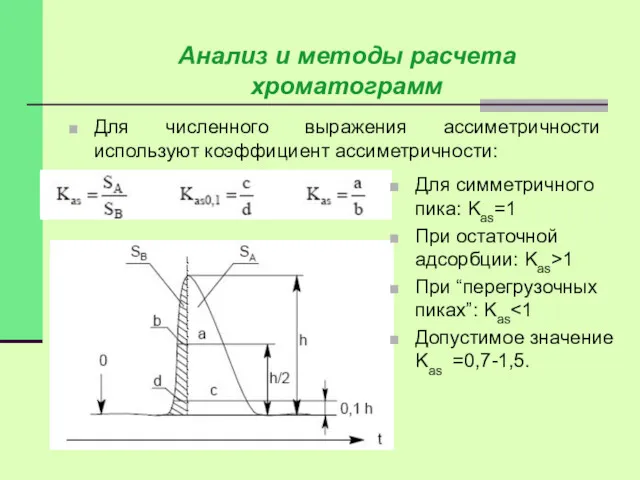
Для численного выражения ассиметричности используют коэффициент ассиметричности:
Анализ и методы расчета хроматограмм
Для
Для численного выражения ассиметричности используют коэффициент ассиметричности:
Анализ и методы расчета хроматограмм
Для
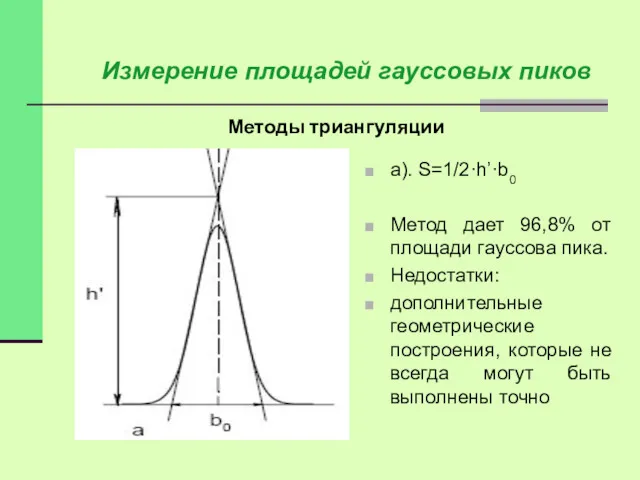
Методы триангуляции
Измерение площадей гауссовых пиков
а). S=1/2·h’·b0
Метод дает 96,8% от площади гауссова
Методы триангуляции
Измерение площадей гауссовых пиков
а). S=1/2·h’·b0
Метод дает 96,8% от площади гауссова
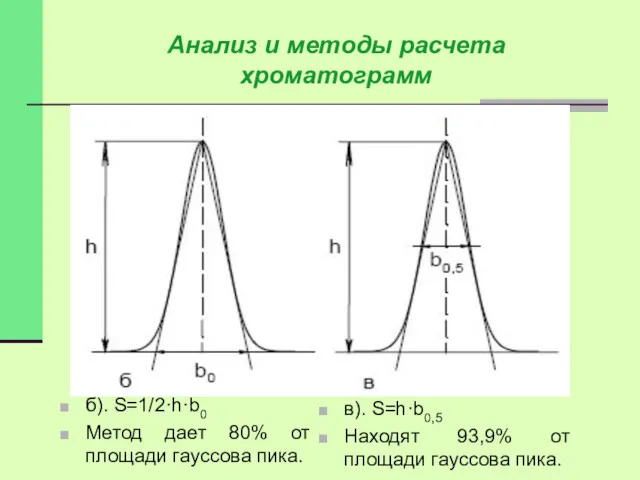
Анализ и методы расчета хроматограмм
б). S=1/2·h·b0
Метод дает 80% от площади гауссова
Анализ и методы расчета хроматограмм
б). S=1/2·h·b0
Метод дает 80% от площади гауссова

Истинная площадь гауссова пика может быть найдена:
Sист=h·b0.368
Анализ и методы расчета хроматограмм
Sист=h·b0.368
Анализ и методы расчета хроматограмм

Количественный состав пробы определяют:
Методом нормировки (внутренней нормализации)
Методом внешней стандартизации (абсолютной градуировки)
Методом
Количественный состав пробы определяют:
Методом нормировки (внутренней нормализации)
Методом внешней стандартизации (абсолютной градуировки)
Методом

Метод нормировки применяется наиболее часто.
Для его применения необходимо, чтобы на
Метод нормировки применяется наиболее часто.
Для его применения необходимо, чтобы на

При анализе смеси трех компонентов, относительное содержание компонентов рассчитывают
Х,% =
При анализе смеси трех компонентов, относительное содержание компонентов рассчитывают
Х,% =

Если чувствительность детектора различна по отношению к каждому компоненту пробы, то
Если чувствительность детектора различна по отношению к каждому компоненту пробы, то

Метод внешней стандартизации (абсолютной градуировки)
Используют при определении отдельных веществ в простых
Метод внешней стандартизации (абсолютной градуировки)
Используют при определении отдельных веществ в простых

Метод внутренней стандартизации применяют при отсутствии на хроматограмме пиков некоторых компонентов
Метод внутренней стандартизации применяют при отсутствии на хроматограмме пиков некоторых компонентов

Сi =k*Сст* Si /Sст;
x% =100 %*k*r* Si /Sст
Сi =k*Сст* Si /Sст;
x% =100 %*k*r* Si /Sст

Качественный анализ
Время удерживания, объем удерживания - характеризуют природу анализируемого вещества
Идентификация по
Качественный анализ
Время удерживания, объем удерживания - характеризуют природу анализируемого вещества
Идентификация по

Идентификация по относительному времени удерживания
Часто для идентификации используют величину относительного
Идентификация по относительному времени удерживания
Часто для идентификации используют величину относительного

Идентификация по индексам удерживания Ковача
За стандарт берут два соседних алкана,
Идентификация по индексам удерживания Ковача
За стандарт берут два соседних алкана,

Идентификация по линейным зависимостям параметров удерживания в гомологическом ряду органических
Идентификация по линейным зависимостям параметров удерживания в гомологическом ряду органических
 Основи молекулярно - кінетичної теорії газів (лекція 6)
Основи молекулярно - кінетичної теорії газів (лекція 6) Что изучает физика
Что изучает физика Теплотехнические схемы парогенераторов АЭС
Теплотехнические схемы парогенераторов АЭС Ремонт системы питания КамАЗ 4326
Ремонт системы питания КамАЗ 4326 Механическое движение. Материальная точка. Система отсчета
Механическое движение. Материальная точка. Система отсчета Типовые детали машин
Типовые детали машин Расчет и проектирование элементов механического измельчения отходов
Расчет и проектирование элементов механического измельчения отходов Молекулярная физика. Молекулярно-кинетическая теория. Масса и размеры молекул
Молекулярная физика. Молекулярно-кинетическая теория. Масса и размеры молекул Равномерное движение по окружности. Решение задач.
Равномерное движение по окружности. Решение задач. Физика пласта. Подземная нефтегидродинамика
Физика пласта. Подземная нефтегидродинамика Исследовательский проект Машина Голдберга Разбивалка яйиц
Исследовательский проект Машина Голдберга Разбивалка яйиц Физические основы механики. Физика в познании вещества, поля, пространства и времени
Физические основы механики. Физика в познании вещества, поля, пространства и времени Звук и его характеристики
Звук и его характеристики Сравнительная характеристика полупроводниковых материалов
Сравнительная характеристика полупроводниковых материалов Общие сведения об устройствах получения информации о процессе
Общие сведения об устройствах получения информации о процессе Lektsia_9_Difraktsia_Frenelya
Lektsia_9_Difraktsia_Frenelya Электромагнитные волны и их свойства. Шкала электромагнитных волн
Электромагнитные волны и их свойства. Шкала электромагнитных волн Озоновый слой атмосферы и озоновые дыры
Озоновый слой атмосферы и озоновые дыры Преломление света. 8 класс
Преломление света. 8 класс Сообщающиеся сосуды
Сообщающиеся сосуды Елементи теорії поля. (Лекція 10)
Елементи теорії поля. (Лекція 10) Материалы с особыми электрическими свойствами
Материалы с особыми электрическими свойствами Процесс ремонтной окраски Mazda 46G
Процесс ремонтной окраски Mazda 46G Дисперсия света
Дисперсия света Методы исследования переключения в сегнетоэлектриках. Микроскопия
Методы исследования переключения в сегнетоэлектриках. Микроскопия Bosch Airless SCR System
Bosch Airless SCR System Электромагнитная совместимость
Электромагнитная совместимость Оптические явления. Урок для 8 класса
Оптические явления. Урок для 8 класса