- Метод молекулярної динаміки

Содержание
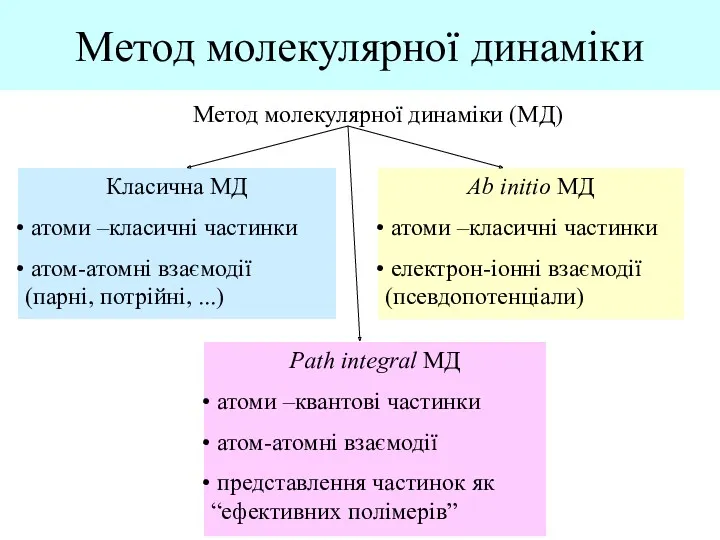
- 2. Метод молекулярної динаміки Метод молекулярної динаміки (МД) Класична МД атоми –класичні частинки атом-атомні взаємодії (парні, потрійні,
- 3. Метод молекулярної динаміки Стандартний алгоритм PROGRAM MD PARAMETER(N=1000,T=600,NSTEP=1000,…..) DIMENSION X(N),Y(N),Z(N) DIMENSION VX(N),VY(N),VZ(N) DIMENSION FX(N),FY(N),FZ(N) CALL INIT
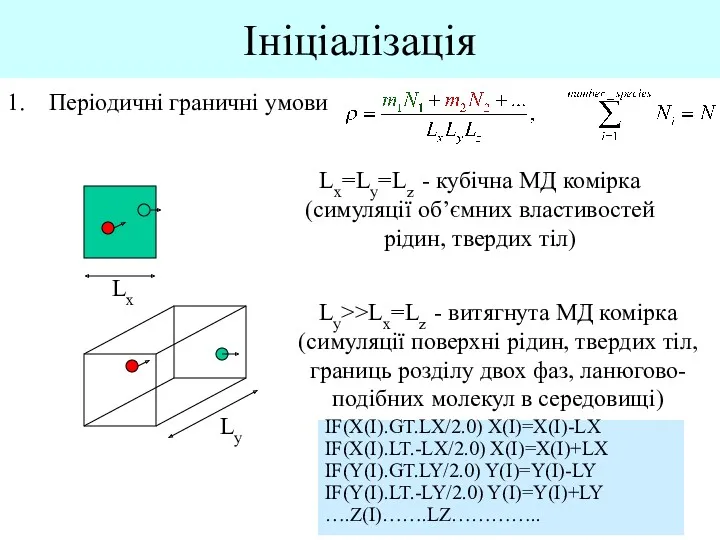
- 4. Ініціалізація Періодичні граничні умови Lx Lx=Ly=Lz - кубічна МД комірка (симуляції об’ємних властивостей рідин, твердих тіл)
- 5. Crystal growth Lennard-Jones (111) solid/liquid interface, T t=600 ps t=100 ps t=300 ps
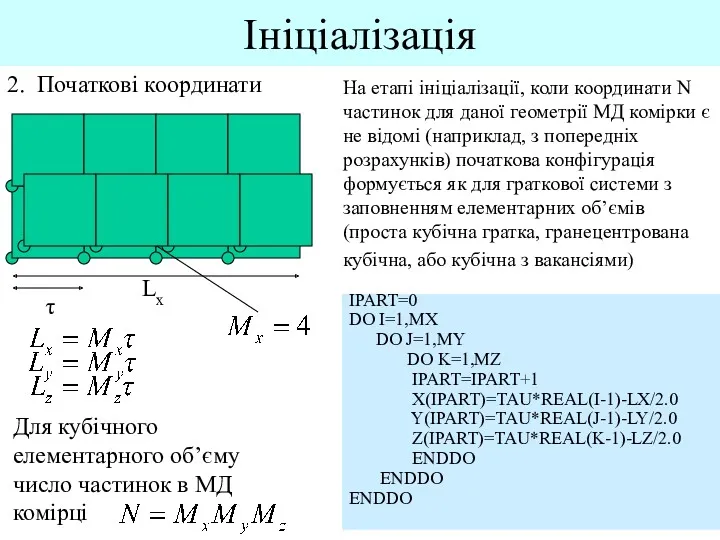
- 6. Ініціалізація 2. Початкові координати Lx На етапі ініціалізації, коли координати N частинок для даної геометрії МД
- 7. Ініціалізація 2. Початкові координати Lx τ Для гранецентрованого кубічного елементарного об’єму число частинок в МД комірці
- 8. Ініціалізація 2. Початкові координати для молекулярних систем Lx τ Для кубічного елементарного об’єму число частинок в
- 9. Ініціалізація 2. Створення нових частинок у вже існуючих конфігураціях Для внесення іона в кристалічну структуру необхідно
- 10. Ініціалізація 3. Початкові швидкості Зв‘язок температури з середньою кінетичною енергією на один ступінь вільності: “Теплова” швидкість:
- 11. Ініціалізація 3. Початкові швидкості Для кожної частинки генеруються з розподілом Максвела x, y, z – компоненти
- 13. Скачать презентацию
Метод молекулярної динаміки
Метод молекулярної динаміки (МД)
Класична МД
атоми –класичні частинки
атом-атомні
Метод молекулярної динаміки
Метод молекулярної динаміки (МД)
Класична МД
атоми –класичні частинки
атом-атомні

Метод молекулярної динаміки
Стандартний алгоритм
PROGRAM MD
PARAMETER(N=1000,T=600,NSTEP=1000,…..)
DIMENSION X(N),Y(N),Z(N)
DIMENSION VX(N),VY(N),VZ(N)
DIMENSION FX(N),FY(N),FZ(N)
CALL INIT (N,T,X,Y,Z,VX,VY,VZ)
DO I=1,NSTEP
Метод молекулярної динаміки
Стандартний алгоритм
PROGRAM MD
PARAMETER(N=1000,T=600,NSTEP=1000,…..)
DIMENSION X(N),Y(N),Z(N)
DIMENSION VX(N),VY(N),VZ(N)
DIMENSION FX(N),FY(N),FZ(N)
CALL INIT (N,T,X,Y,Z,VX,VY,VZ)
DO I=1,NSTEP
Ініціалізація
Періодичні граничні умови
Lx
Lx=Ly=Lz - кубічна МД комірка (симуляції об’ємних властивостей рідин,
Ініціалізація
Періодичні граничні умови
Lx
Lx=Ly=Lz - кубічна МД комірка (симуляції об’ємних властивостей рідин,
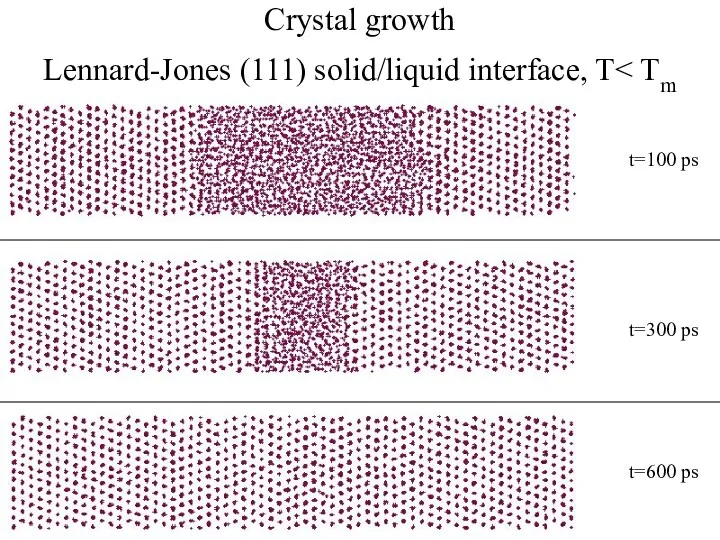
Crystal growth
Lennard-Jones (111) solid/liquid interface, T< Tm
t=600 ps
Crystal growth
Lennard-Jones (111) solid/liquid interface, T< Tm
t=600 ps
Ініціалізація
2. Початкові координати
Lx
На етапі ініціалізації, коли координати N частинок для даної
Ініціалізація
2. Початкові координати
Lx
На етапі ініціалізації, коли координати N частинок для даної
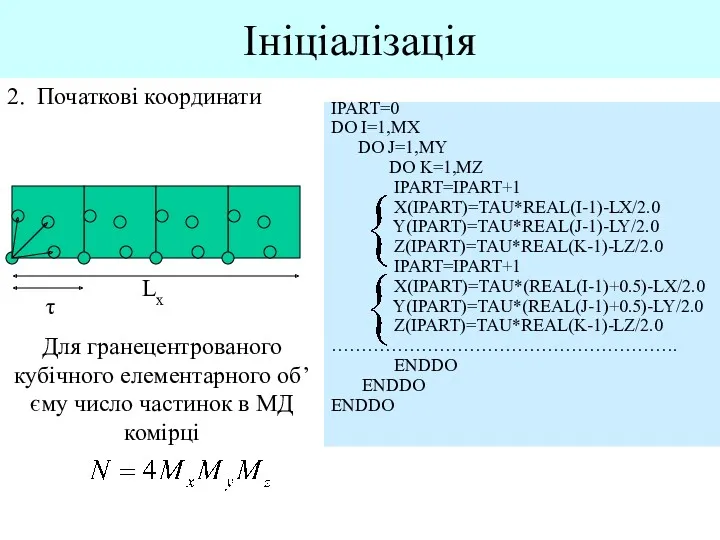
Ініціалізація
2. Початкові координати
Lx
τ
Для гранецентрованого кубічного елементарного об’єму число частинок в МД
Ініціалізація
2. Початкові координати
Lx
τ
Для гранецентрованого кубічного елементарного об’єму число частинок в МД
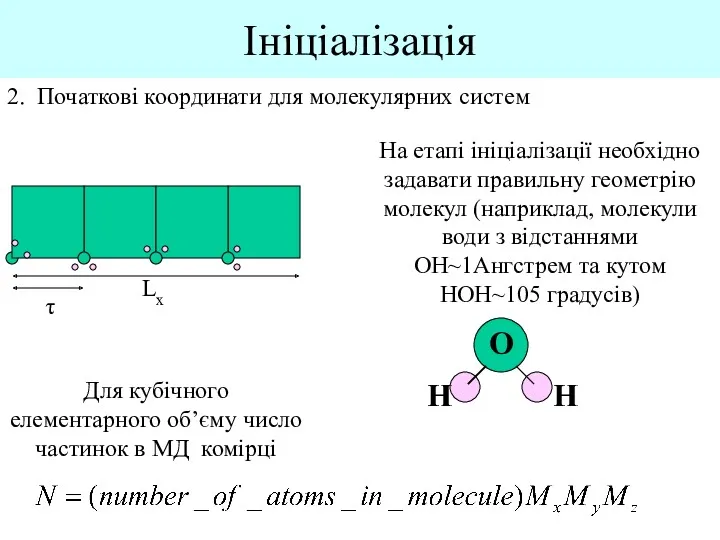
Ініціалізація
2. Початкові координати для молекулярних систем
Lx
τ
Для кубічного елементарного об’єму число частинок
Ініціалізація
2. Початкові координати для молекулярних систем
Lx
τ
Для кубічного елементарного об’єму число частинок
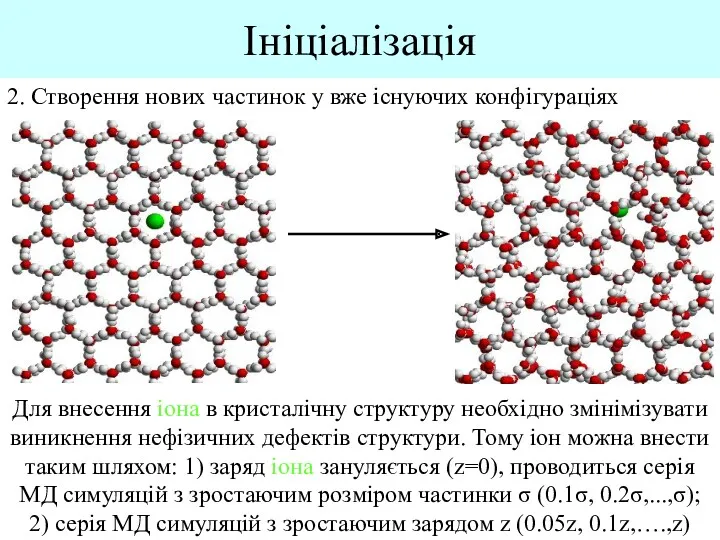
Ініціалізація
2. Створення нових частинок у вже існуючих конфігураціях
Для внесення іона в
Ініціалізація
2. Створення нових частинок у вже існуючих конфігураціях
Для внесення іона в
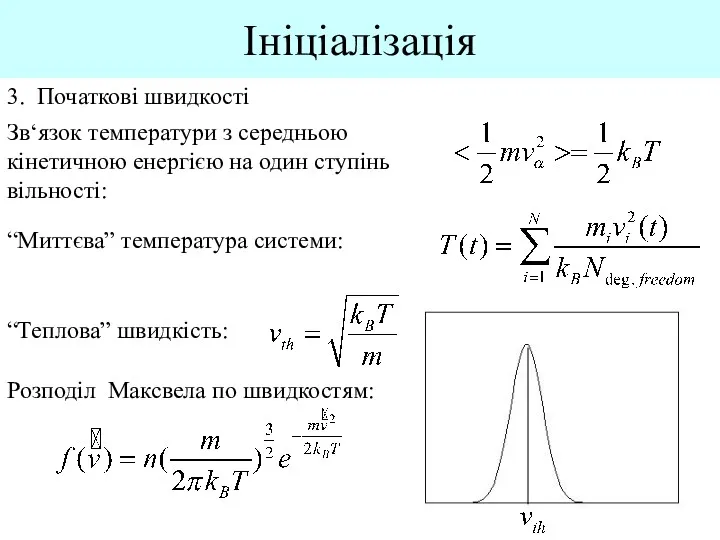
Ініціалізація
3. Початкові швидкості
Зв‘язок температури з середньою кінетичною енергією на один ступінь
Ініціалізація
3. Початкові швидкості
Зв‘язок температури з середньою кінетичною енергією на один ступінь

Ініціалізація
3. Початкові швидкості
Для кожної частинки генеруються з розподілом Максвела x, y,
Ініціалізація
3. Початкові швидкості
Для кожної частинки генеруються з розподілом Максвела x, y,
 Исследовательский проект Внедрение QR-кодов в различные сферы нашей жизни
Исследовательский проект Внедрение QR-кодов в различные сферы нашей жизни Основы веб-разработки. HTML. Изображения, видео, звук. (Лекция 5)
Основы веб-разработки. HTML. Изображения, видео, звук. (Лекция 5) Решаемые задачи для государства
Решаемые задачи для государства Конструирование программного обеспечения. Контейнеры и коллекции объектов
Конструирование программного обеспечения. Контейнеры и коллекции объектов An Exercise of SQL Using SQL* Plus
An Exercise of SQL Using SQL* Plus Презентация Файловые архивы
Презентация Файловые архивы Итоги 2017 года отдела по работе с интернет-магазинами
Итоги 2017 года отдела по работе с интернет-магазинами Построение диаграмм и графиков в электронных таблицах
Построение диаграмм и графиков в электронных таблицах Обзор информационных ресурсов для изучения английского языка
Обзор информационных ресурсов для изучения английского языка Types of computer programs, and their use
Types of computer programs, and their use Векторная графика. Графические возможности MS Word
Векторная графика. Графические возможности MS Word Системный анализ: Кибернетические модели. Экономико-математические модели. Нормативные операционные модели
Системный анализ: Кибернетические модели. Экономико-математические модели. Нормативные операционные модели WEB-Index: Аудитория интернет-проектов. Desktop, Октябрь 2017, Москва
WEB-Index: Аудитория интернет-проектов. Desktop, Октябрь 2017, Москва Жаңа форматтағы ҰБТ/КТ-ге дайындалуға арналған онлайн байқау сынағын өткізуге арналған портал
Жаңа форматтағы ҰБТ/КТ-ге дайындалуға арналған онлайн байқау сынағын өткізуге арналған портал Глобальна інформаційна інфраструктура. (Лекція 1)
Глобальна інформаційна інфраструктура. (Лекція 1) Кирилл Соеров (популярный анимешный видео-блогер, в своих видео он делает обзор на разные аниме)
Кирилл Соеров (популярный анимешный видео-блогер, в своих видео он делает обзор на разные аниме) Влияние социальных сетей на грамматность речи
Влияние социальных сетей на грамматность речи Интерактивные доски
Интерактивные доски Алгоритми на графах 1
Алгоритми на графах 1 В контакте. Создание и ведение сообщества в ВК
В контакте. Создание и ведение сообщества в ВК Базы данных
Базы данных Архитектура Oracle. Программные модули (PL/SQL, лекция 12)
Архитектура Oracle. Программные модули (PL/SQL, лекция 12) Программирование графических объектов в среде Pascal ABC
Программирование графических объектов в среде Pascal ABC Інформаційна асиметрія та реклама
Інформаційна асиметрія та реклама Типы и структура уроков по ФГОС
Типы и структура уроков по ФГОС Робототехника. Сборка к занятию №5. Изучение ременной передачи
Робототехника. Сборка к занятию №5. Изучение ременной передачи Обработка информации и алгоритмы
Обработка информации и алгоритмы Теория информации. Теорема кодирования
Теория информации. Теорема кодирования