- Аномалии развития мозжечка и мозолистого тела

Содержание
- 2. Гипоплазия мозжечка Данным термином объединяется большая группа атактических синдромов, характеризующихся нарушением нормального отногенетического развития и дифференцировки
- 3. Классификация форм гипоплазии мозжечка (Patel S, Barkovich AJ, 2002): A.Фокальная гипоплазия 1.Изолированная гипоплазия червя 2.Изолированная гипоплазия
- 4. Генерализованная гипоплазия с расширением IV желудочка (Dandy—Walker Continuum). Аномалия структур задней черепной ямки была впервые описана
- 5. Аномалия Денди–Уокера (DWM) представляет собой врожденный порок развития крыши IVжелудочка и червя мозжечка, ведущий к неполному
- 6. 1. характерная триада при Dandy-Walker Malformation: гипоплазия червя мозжечка (vermis), кистообразное расширение IV желудочка (4V), расширенная
- 7. Клинические проявления аномалии Денди–Уокера включают задержку психомоторного развития, макроцефалию, гидроцефалию различной степени выраженности (Niesen CE et
- 8. Известна роль мозжечка в контроле и интеграции моторной деятельности. Однако, его многочисленные связи с ассоциативными и
- 9. Генерализованная церебеллярная гипоплазия с нормальным IV желудочком В классификации аномалий мозжечка, предложенной Patel S и Barkovich
- 10. 1. сагиттальное T1-weighted изображение, демонстрирует генерализованную гипоплазию мозжечка при нормальных размерах задней черепной ямки ассоциированную с
- 11. X-сцепленная церебеллярная гипоплазия Фенотипические проявления синдрома включают макроцефалию, диспластичное лицо, высокий рост, гипогенитализм. Клинические признаки заболевания
- 12. PHACE syndrome Церебеллярная гипоплазия с гемангиомой лица впервые описана Pascual-Castroviejo I (1978). В 1981г. Sawaya и
- 13. Сагиттальное и фронтальное T1-weighted изображение асимметричной гипоплазии червя и полушарий мозжечка при нормальных размерах задней черепной
- 14. Ritscher–Schinzel syndrome. Синдром был описан у двух сестёр Ritscher и соавторами в 1987 году. Одна из
- 15. Hoyeraal–Hreidarsson syndrome. В 1988 году Hreidarsson с соавторами описали мальчика умершего в возрасте 23 месяцев. Он
- 16. Врождённая непрогрессирующая атаксия. К данной группе относятся пациенты, не имеющие признаков инфекционного поражения ЦНС, перенесённого перинатального
- 17. В соответствии с классификацией Barkovich AJ. (2002) К группе генерализованных церебеллярных гипоплазий с нормальным IV желудочком
- 18. 23.2.16 В 1995 году Barth et al. выделил два типа патологии: Тип I (PCHI) сопровождался дегенеративными
- 19. Генерализованная гипоплазия мозжечка характерна для спинальной мышечной атрофии с понтоцеребеллярной гипоплазией. Данное заболевание трудно для дифференциальной
- 20. Фокальная гипоплазия мозжечка Изолированная гипоплазия одной гемисферы. Первое описание унилатеральной церебеллярной гипоплазии датируется 1915 годом, когда
- 21. Аксиальное T1-weighted изображение, демонстрирует гипоплазию правой гемисферы мозжечка при нормальных размерах червя и правой гемисферы (Patel
- 22. Изолированная гипоплазия червя мозжечка. Изолированная гипоплазия нижних отделов червя мозжечка, не всегда рассматривается как аномалия (Patel
- 23. Считают, что существует связь между патологией мозжечка и эпилептическим приступом. Мозжечок – практически единственная структура, в
- 24. 23.2.16 Роль мозжечка в развитии эпилепсии. Проф. Stefan Herlitze с соавт., (Рурский университет, Германия), показали, что
- 25. Агенезия мозолистого тела. В основе патологии лежит нарушение дифференциации комиссуральной пластинки в период между 12-й и
- 26. 23.2.16
- 27. 23.2.16 При полной (тотальной) агенезии клинически могут отмечаться дисморфические признаки (макроцефалия, гипертелоризм), мышечная гипотония, задержка умственного
- 28. 23.2.16 Агенезия мозолистого тела легко распознаваема методами нейровизуализации. Определяется увеличенный вперед и вверх III желудочек, который
- 31. 23.2.16
- 32. 23.2.16
- 33. Пренатальная диагностика агенезии мозолистого тела достаточно сложна. Основные сроки выявления данной патологии – II, III триместр
- 34. 23.2.16 Срок беременности 32 недели. МРТ Т2 ВИ. Коронарная плоскость. Мозолистое тело отсутствует. Расстояние между телами
- 35. 23.2.16
- 36. 23.2.16 Мозг новорожденного в отличие от мозга взрослого человека очень «пластичен». В нем еще не произошло
- 38. Скачать презентацию

Гипоплазия мозжечка
Данным термином объединяется большая группа атактических синдромов, характеризующихся нарушением нормального
Гипоплазия мозжечка
Данным термином объединяется большая группа атактических синдромов, характеризующихся нарушением нормального

Классификация форм гипоплазии мозжечка (Patel S, Barkovich AJ, 2002):
A.Фокальная гипоплазия
1.Изолированная гипоплазия червя
2.Изолированная гипоплазия одной
Классификация форм гипоплазии мозжечка (Patel S, Barkovich AJ, 2002):
A.Фокальная гипоплазия
1.Изолированная гипоплазия червя
2.Изолированная гипоплазия одной

Генерализованная гипоплазия с расширением IV желудочка (Dandy—Walker Continuum).
Аномалия структур задней черепной ямки была впервые
Генерализованная гипоплазия с расширением IV желудочка (Dandy—Walker Continuum).
Аномалия структур задней черепной ямки была впервые

Аномалия Денди–Уокера (DWM) представляет собой врожденный порок развития крыши IVжелудочка и червя
Аномалия Денди–Уокера (DWM) представляет собой врожденный порок развития крыши IVжелудочка и червя
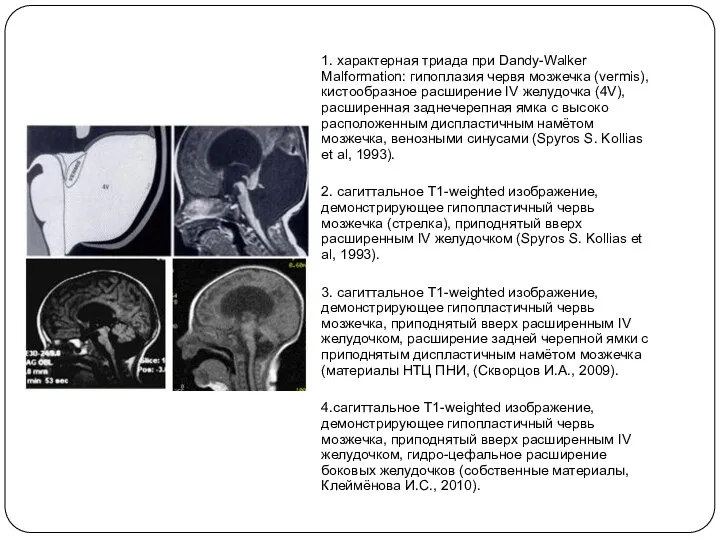
1. характерная триада при Dandy-Walker Malformation: гипоплазия червя мозжечка (vermis), кистообразное
1. характерная триада при Dandy-Walker Malformation: гипоплазия червя мозжечка (vermis), кистообразное

Клинические проявления аномалии Денди–Уокера включают задержку психомоторного развития, макроцефалию, гидроцефалию различной
Клинические проявления аномалии Денди–Уокера включают задержку психомоторного развития, макроцефалию, гидроцефалию различной

Известна роль мозжечка в контроле и интеграции моторной деятельности. Однако, его
Известна роль мозжечка в контроле и интеграции моторной деятельности. Однако, его

Генерализованная церебеллярная гипоплазия с нормальным IV желудочком
В классификации аномалий мозжечка, предложенной
Генерализованная церебеллярная гипоплазия с нормальным IV желудочком
В классификации аномалий мозжечка, предложенной
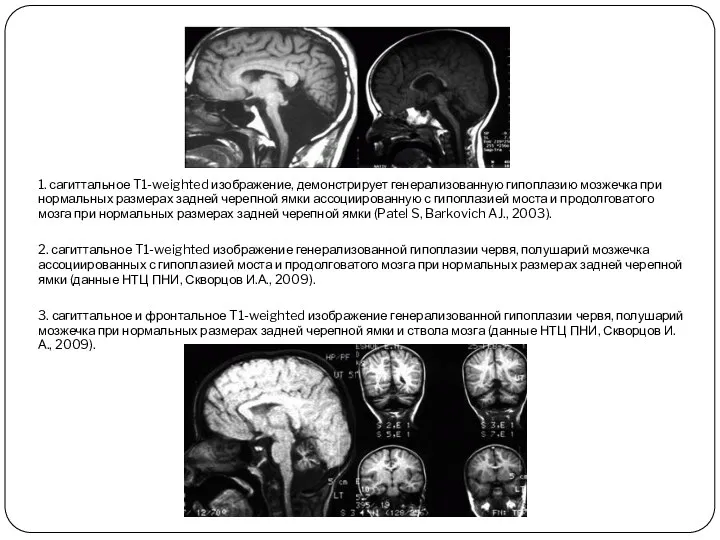
1. сагиттальное T1-weighted изображение, демонстрирует генерализованную гипоплазию мозжечка при нормальных размерах
1. сагиттальное T1-weighted изображение, демонстрирует генерализованную гипоплазию мозжечка при нормальных размерах

X-сцепленная церебеллярная гипоплазия
Фенотипические проявления синдрома включают макроцефалию, диспластичное лицо, высокий
X-сцепленная церебеллярная гипоплазия
Фенотипические проявления синдрома включают макроцефалию, диспластичное лицо, высокий

PHACE syndrome
Церебеллярная гипоплазия с гемангиомой лица впервые описана Pascual-Castroviejo I (1978).
PHACE syndrome
Церебеллярная гипоплазия с гемангиомой лица впервые описана Pascual-Castroviejo I (1978).
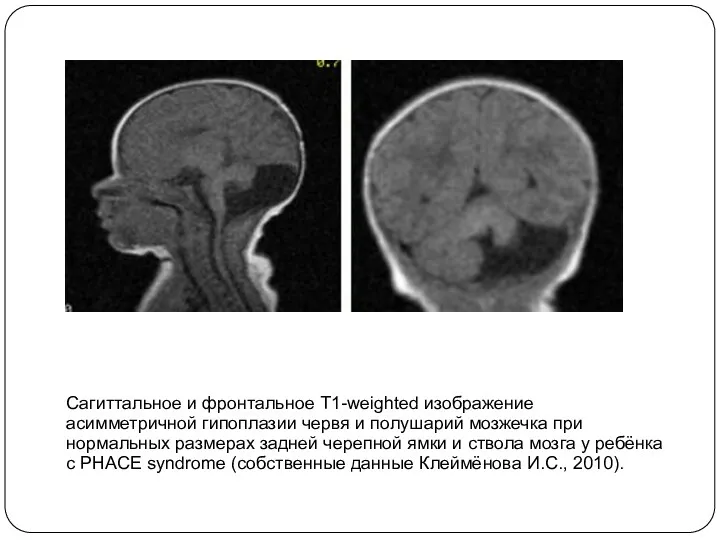
Сагиттальное и фронтальное T1-weighted изображение асимметричной гипоплазии червя и полушарий мозжечка
Сагиттальное и фронтальное T1-weighted изображение асимметричной гипоплазии червя и полушарий мозжечка

Ritscher–Schinzel syndrome.
Синдром был описан у двух сестёр Ritscher и соавторами
Ritscher–Schinzel syndrome.
Синдром был описан у двух сестёр Ritscher и соавторами

Hoyeraal–Hreidarsson syndrome.
В 1988 году Hreidarsson с соавторами описали мальчика умершего
Hoyeraal–Hreidarsson syndrome.
В 1988 году Hreidarsson с соавторами описали мальчика умершего

Врождённая непрогрессирующая атаксия.
К данной группе относятся пациенты, не имеющие признаков
Врождённая непрогрессирующая атаксия.
К данной группе относятся пациенты, не имеющие признаков

В соответствии с классификацией Barkovich AJ. (2002) К группе генерализованных церебеллярных
В соответствии с классификацией Barkovich AJ. (2002) К группе генерализованных церебеллярных

23.2.16
В 1995 году Barth et al. выделил два типа патологии:
Тип I
23.2.16
В 1995 году Barth et al. выделил два типа патологии:
Тип I

Генерализованная гипоплазия мозжечка характерна для спинальной мышечной атрофии с понтоцеребеллярной гипоплазией.
Генерализованная гипоплазия мозжечка характерна для спинальной мышечной атрофии с понтоцеребеллярной гипоплазией.

Фокальная гипоплазия мозжечка
Изолированная гипоплазия одной гемисферы.
Первое описание унилатеральной церебеллярной гипоплазии
Фокальная гипоплазия мозжечка
Изолированная гипоплазия одной гемисферы.
Первое описание унилатеральной церебеллярной гипоплазии
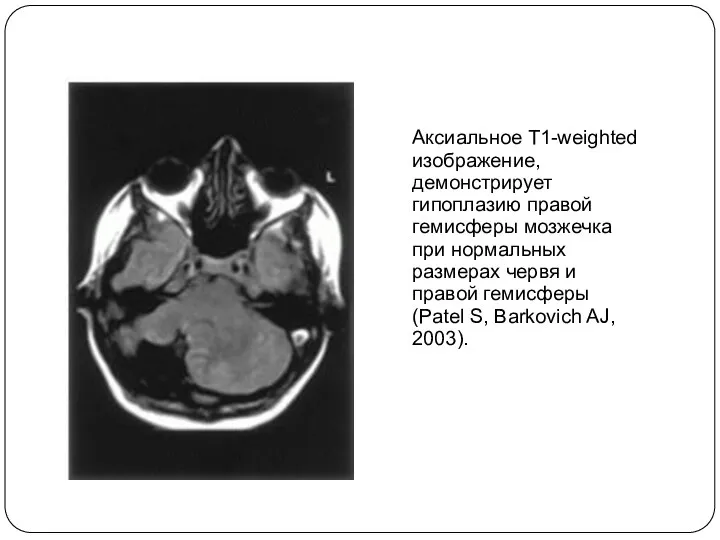
Аксиальное T1-weighted изображение, демонстрирует гипоплазию правой гемисферы мозжечка при нормальных размерах
Аксиальное T1-weighted изображение, демонстрирует гипоплазию правой гемисферы мозжечка при нормальных размерах

Изолированная гипоплазия червя мозжечка.
Изолированная гипоплазия нижних отделов червя мозжечка, не
Изолированная гипоплазия червя мозжечка.
Изолированная гипоплазия нижних отделов червя мозжечка, не

Считают, что существует связь между патологией мозжечка и эпилептическим приступом.
Мозжечок –
Считают, что существует связь между патологией мозжечка и эпилептическим приступом.
Мозжечок –

23.2.16
Роль мозжечка в развитии эпилепсии.
Проф. Stefan Herlitze с соавт., (Рурский университет,
23.2.16
Роль мозжечка в развитии эпилепсии.
Проф. Stefan Herlitze с соавт., (Рурский университет,
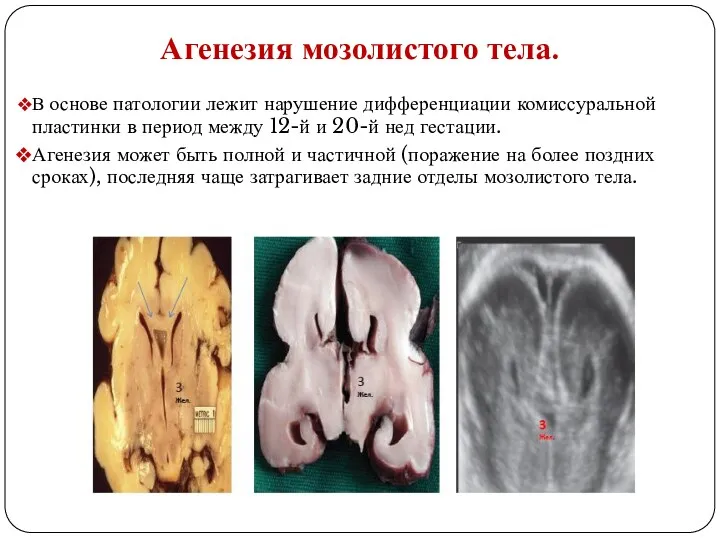
Агенезия мозолистого тела.
В основе патологии лежит нарушение дифференциации комиссуральной пластинки
Агенезия мозолистого тела.
В основе патологии лежит нарушение дифференциации комиссуральной пластинки

23.2.16
23.2.16

23.2.16
При полной (тотальной) агенезии клинически могут отмечаться дисморфические признаки (макроцефалия, гипертелоризм),
23.2.16
При полной (тотальной) агенезии клинически могут отмечаться дисморфические признаки (макроцефалия, гипертелоризм),

23.2.16
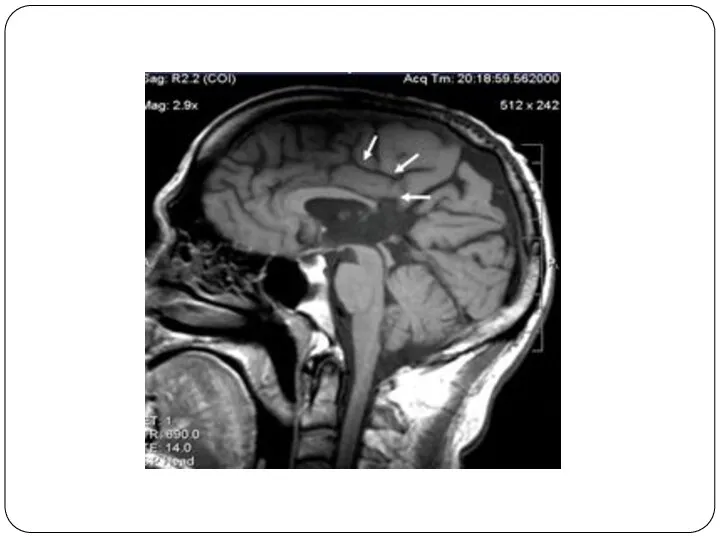
Агенезия мозолистого тела легко распознаваема методами нейровизуализации.
Определяется увеличенный вперед и
23.2.16
Агенезия мозолистого тела легко распознаваема методами нейровизуализации.
Определяется увеличенный вперед и
23.2.16
23.2.16
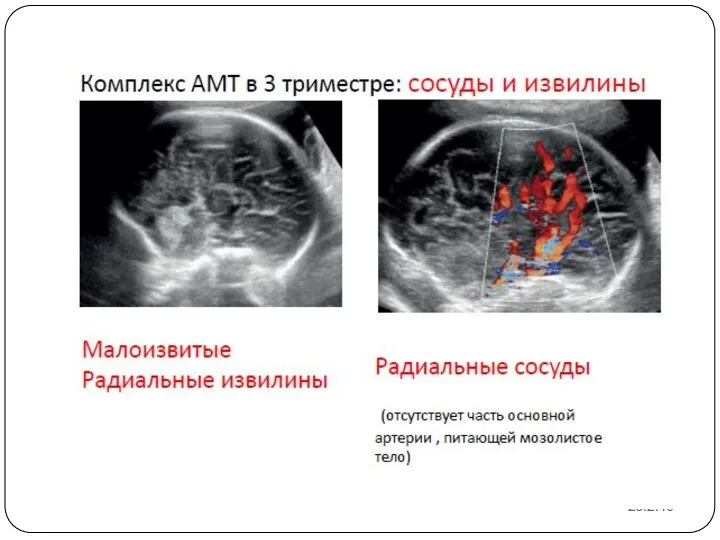
23.2.16
23.2.16

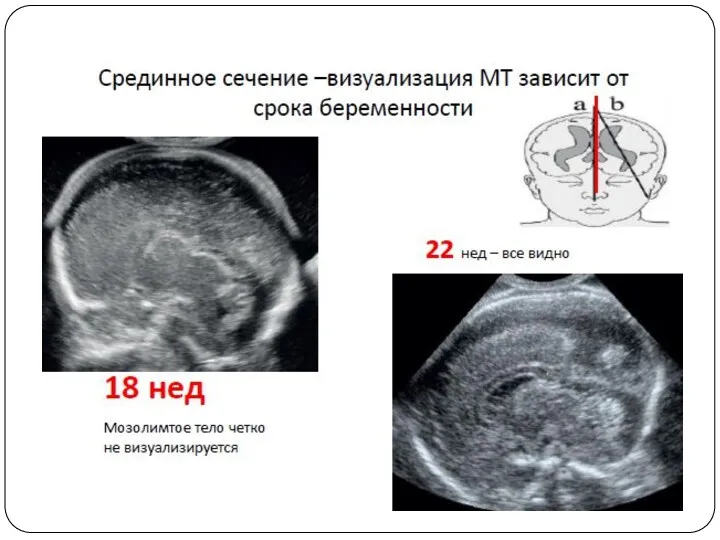
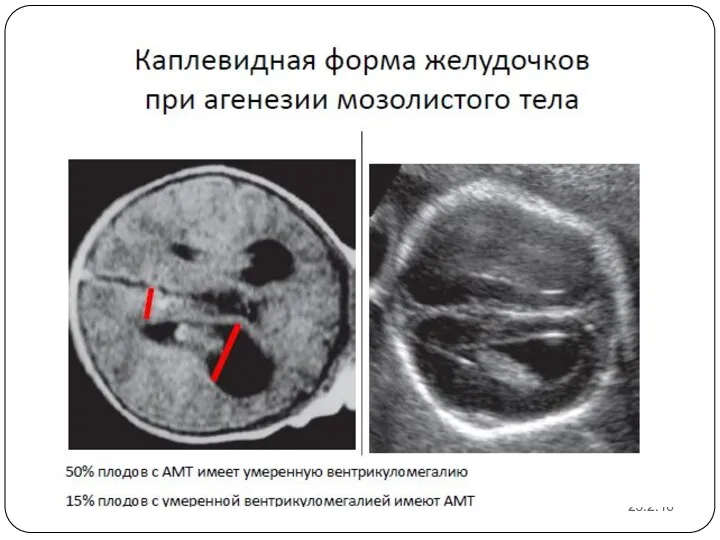
Пренатальная диагностика агенезии мозолистого тела достаточно сложна. Основные сроки выявления данной
Пренатальная диагностика агенезии мозолистого тела достаточно сложна. Основные сроки выявления данной
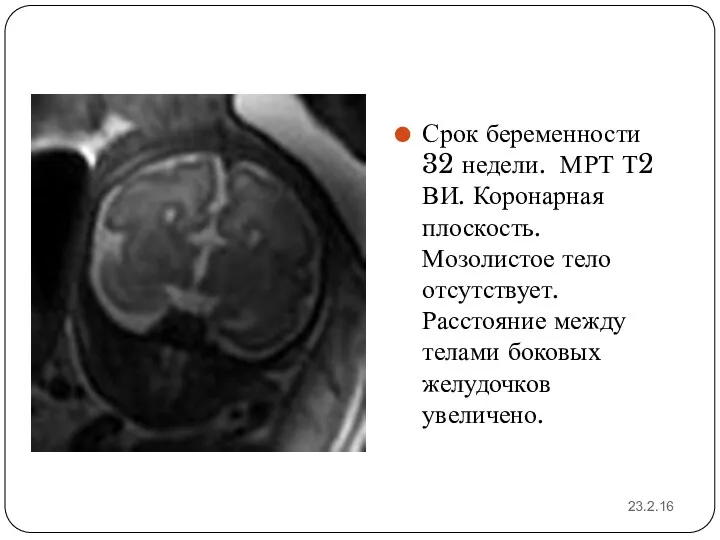
23.2.16
Срок беременности 32 недели. МРТ Т2 ВИ. Коронарная плоскость. Мозолистое тело
23.2.16
Срок беременности 32 недели. МРТ Т2 ВИ. Коронарная плоскость. Мозолистое тело

23.2.16
23.2.16

23.2.16
Мозг новорожденного в отличие от мозга взрослого человека очень «пластичен». В
23.2.16
Мозг новорожденного в отличие от мозга взрослого человека очень «пластичен». В
 Клуб правильного питания. Часть 1
Клуб правильного питания. Часть 1 Вроджені вади грудної клітки
Вроджені вади грудної клітки Эпидемиология и профилактика холеры
Эпидемиология и профилактика холеры Обоснование применения визуальных методов исследования в гинекологии
Обоснование применения визуальных методов исследования в гинекологии Балалардағы эндокриндік патологияның визуалді диагностика әдістерінің ерекшеліктері
Балалардағы эндокриндік патологияның визуалді диагностика әдістерінің ерекшеліктері Тактические принципы работы выездных бригад скорой медицинской помощи
Тактические принципы работы выездных бригад скорой медицинской помощи Сальмонеллёзы. Этиология
Сальмонеллёзы. Этиология Инфекция. Роль микроорганизмов в развитии инфекционных заболеваний человека
Инфекция. Роль микроорганизмов в развитии инфекционных заболеваний человека Анализ ассортимента антиаритмических препаратов в аптечной организации
Анализ ассортимента антиаритмических препаратов в аптечной организации Жүрекшелер фибрилляция кезінде анти аритмиялық препараттарды (амидарон, флекайнид) салыстыру
Жүрекшелер фибрилляция кезінде анти аритмиялық препараттарды (амидарон, флекайнид) салыстыру Совершенствование организационной структуры медицинской организации (на примере ООО ММЦ Диалайн)
Совершенствование организационной структуры медицинской организации (на примере ООО ММЦ Диалайн) Ісік жасушаларын зерттеу
Ісік жасушаларын зерттеу Раневые инфекции. Этиологическая структура
Раневые инфекции. Этиологическая структура Современные принципы организации акушерскогинекологической помощи
Современные принципы организации акушерскогинекологической помощи Презентация ООИБ-ПНД
Презентация ООИБ-ПНД Клиника и лечение современной холеры
Клиника и лечение современной холеры Страховая модель здравоохранения
Страховая модель здравоохранения Жүре пайда болған жүрек ақаулары
Жүре пайда болған жүрек ақаулары Назальный глюкагон как альтернатива введению глюкагона у детей с сахарным диабетом I типа при гипогликемических состояниях
Назальный глюкагон как альтернатива введению глюкагона у детей с сахарным диабетом I типа при гипогликемических состояниях Профилактика врожденной патологии лица
Профилактика врожденной патологии лица Опухолевый рост
Опухолевый рост Интерннің өзіндік жұмысы
Интерннің өзіндік жұмысы Гематогенді остеомиелит
Гематогенді остеомиелит Симптоматическая артериальная гипертензия
Симптоматическая артериальная гипертензия Эпилепсия. Классификация припадков. Первая помощь
Эпилепсия. Классификация припадков. Первая помощь Правильная осанка
Правильная осанка Диссеминированное внутрисосудистое свертывание крови (ДВС)
Диссеминированное внутрисосудистое свертывание крови (ДВС) Интерферон короткого действия
Интерферон короткого действия