- Державна реєстрація лікарських засобів, мікробіологічних та імуноферментних препаратів

Содержание
- 2. Державне підприємство “Державний експертний центр Міністерства охорони здоров’я України” - уповноважена Міністерством охорони здоров'я України спеціалізована
- 3. Накази МОЗ якими регулюється процес реєстрації лікарських засобів в Україні 1. Наказ МОЗ № 426 від
- 4. Порядок реєстрації ЛЗ та МІБП затверджений наказом МОЗ України №460 не поширюються, на: препарати на основі
- 5. Порядок поширюється на препарати отримані з людської крові або плазми. Відповідно п.32 розділу ІІ лікарські засоби,
- 6. При проведенні реєстрації препаратів отриманих з людської крові або плазми, виробник зобов'язаний довести свою здатність досягти
- 7. Реєстрація препаратів крові в Євросоюзі: Збір та дослідження крові та її компонентів регулюється
- 8. Реєстрація препаратів крові в Євросоюзі: збір та дослідження крові та її компонентів регулюється
- 9. Збір та дослідження крові та її компонентів регулюється
- 10. Вимоги до зберігання, транспортування та дистрибюції крові та її компонентів відповідно до Директиви 2004/33 / ЄC
- 11. Збір плази для фракціонування: Керівництво ЄС з Належної виробничої практики (GMP) (http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-4/annex14_rev30-03_2011_en.pdf) Керівництво ЄС щодо вимог
- 12. Керівництво ЄС з Належної виробничої практики (GMP) (http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-4/annex14_rev30-03_2011_en.pdf): додаток 14: якщо плазма постачається до ЄС з
- 14. Скачать презентацию

Державне підприємство
“Державний експертний центр
Міністерства охорони здоров’я України”
- уповноважена
Державне підприємство
“Державний експертний центр
Міністерства охорони здоров’я України”
- уповноважена

Накази МОЗ якими регулюється процес
реєстрації лікарських засобів в Україні
1. Наказ
Накази МОЗ якими регулюється процес
реєстрації лікарських засобів в Україні
1. Наказ

Порядок реєстрації ЛЗ та МІБП
затверджений наказом МОЗ України №460
не
Порядок реєстрації ЛЗ та МІБП
затверджений наказом МОЗ України №460
не

Порядок поширюється на препарати
отримані з людської крові або плазми.
Відповідно
Порядок поширюється на препарати
отримані з людської крові або плазми.
Відповідно

При проведенні реєстрації препаратів отриманих з людської крові або плазми, виробник
При проведенні реєстрації препаратів отриманих з людської крові або плазми, виробник

Реєстрація препаратів крові в Євросоюзі:
Збір та дослідження крові та її компонентів
Реєстрація препаратів крові в Євросоюзі:
Збір та дослідження крові та її компонентів

Реєстрація препаратів крові в Євросоюзі:
збір та дослідження крові та її
Реєстрація препаратів крові в Євросоюзі:
збір та дослідження крові та її

Збір та дослідження крові та її компонентів регулюється
Збір та дослідження крові та її компонентів регулюється

Вимоги до зберігання, транспортування та дистрибюції крові та її компонентів відповідно
Вимоги до зберігання, транспортування та дистрибюції крові та її компонентів відповідно

Збір плази для фракціонування:
Керівництво ЄС з Належної виробничої практики (GMP)
Збір плази для фракціонування:
Керівництво ЄС з Належної виробничої практики (GMP)

Керівництво ЄС з Належної виробничої практики (GMP) (http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-4/annex14_rev30-03_2011_en.pdf):
додаток 14: якщо плазма
Керівництво ЄС з Належної виробничої практики (GMP) (http://ec.europa.eu/health//sites/health/files/files/eudralex/vol-4/annex14_rev30-03_2011_en.pdf):
додаток 14: якщо плазма
 Клиническая анатомия конечностей. Современная ангиология
Клиническая анатомия конечностей. Современная ангиология Аритмиялар (өміріне қауіп төндірген аритмия кезінде ауруханаға жатқызғанға дейін шұғыл және жедел көмек көрсету)
Аритмиялар (өміріне қауіп төндірген аритмия кезінде ауруханаға жатқызғанға дейін шұғыл және жедел көмек көрсету) Guidelines for the use of antiretroviral agents in adults and adolescents
Guidelines for the use of antiretroviral agents in adults and adolescents Катаракта. Причины катаракты
Катаракта. Причины катаракты Сүйек және буын туберкулезі
Сүйек және буын туберкулезі Повреждение костей таза и тазовых органов
Повреждение костей таза и тазовых органов Профилактика внутрибольничных инфекций, связанных и оказанием медицинской помощи
Профилактика внутрибольничных инфекций, связанных и оказанием медицинской помощи Рахит. Этиопатогенез рахита
Рахит. Этиопатогенез рахита Урология. Несеп-жыныс жүйесінің жарақаты
Урология. Несеп-жыныс жүйесінің жарақаты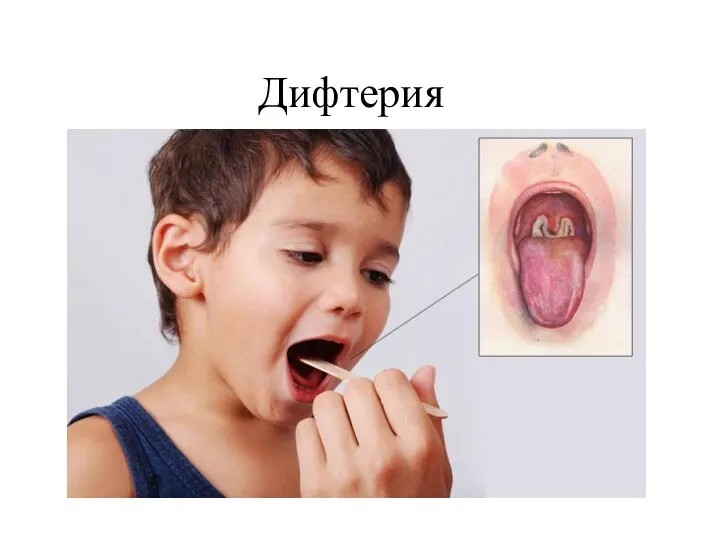 Дифтерия: этиология, классификация, клиника, лечение
Дифтерия: этиология, классификация, клиника, лечение Сестринский уход при сахарном диабете. Часть 1
Сестринский уход при сахарном диабете. Часть 1 Реакция отторжения трансплантата
Реакция отторжения трансплантата Острые тромбозы и эмболии аорты и магистральных артерий
Острые тромбозы и эмболии аорты и магистральных артерий Патологиялық анатомия. Кардиосклероз
Патологиялық анатомия. Кардиосклероз Табиғи ошақты және антропозоонозды инвазиялар
Табиғи ошақты және антропозоонозды инвазиялар Перевязочные материалы. Виды, потребительские свойства, оценка качества
Перевязочные материалы. Виды, потребительские свойства, оценка качества Лекарственные растения. Берёзовая роща
Лекарственные растения. Берёзовая роща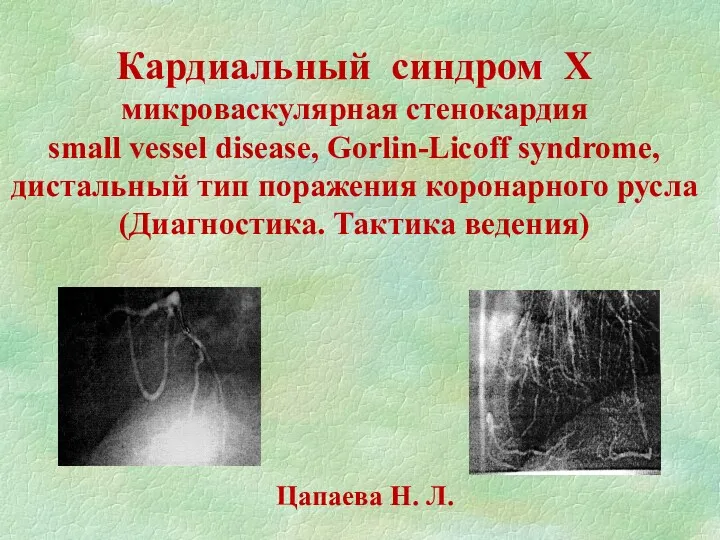 Кардиальный синдром Х. Микроваскулярная стенокардия small vessel disease, дистальный тип поражения коронарного русла
Кардиальный синдром Х. Микроваскулярная стенокардия small vessel disease, дистальный тип поражения коронарного русла Актиномицеты сем. Actinomycetaceae
Актиномицеты сем. Actinomycetaceae Основные клинико-лабораторные методы оценки качества зубных щеток
Основные клинико-лабораторные методы оценки качества зубных щеток Отечественный опыт медицинского обслуживания международных спортивных мероприятий на примере Республики Татарстан
Отечественный опыт медицинского обслуживания международных спортивных мероприятий на примере Республики Татарстан Особенности этиопатогенетических факторов у пациентов с различными видами нехимических зависимостей
Особенности этиопатогенетических факторов у пациентов с различными видами нехимических зависимостей Introduction about ticks
Introduction about ticks Иммунопатология
Иммунопатология Вакцинация и ревакцинация. Вакцина БЦЖ
Вакцинация и ревакцинация. Вакцина БЦЖ Артериалды гипертензия
Артериалды гипертензия Мочевой синдром
Мочевой синдром 16_Polovye_gormony
16_Polovye_gormony