- Дифференциальная диагностика прионных болезней

Содержание
- 2. Типы прионных болезней с учётом причины их возникновения
- 3. ВОЗ была предложена классификация, включающая 3 основных типа развития БКЯ: I тип Нарушения зрения, прогрессирующая деменция,
- 4. Эпидемиологические особенности Убиквитарный (повсеместно) характер распространения заболеваний (некоторые европейские страны, Австралия, страны Северной и Южной Америки)
- 5. В эпидемиологическом отношении БКЯ может проявляться в спорадической, семейной (наследственной) и ятрогенной формах Ганс Герхард Крейтцфельдт
- 6. Куру является эндемической медленной инфекцией, встречающейся в восточной части о. Новая Гвинея и выявлена у племен
- 7. 1. Неврологические осложнения при системных васкулитах Васкулит – это аутоиммунное воспаление стенки сосуда (артерии, вены, артериолы,
- 8. 2.Антифосфолипидный синдром (АФС) это аутоиммунное заболевание, преимущественно женщин молодого возраста, характеризующееся клинико-лабораторным симптомокомплексом, включающим рецидивирующие тромбозы,
- 9. Антифосфолипидный синдром (АФС) – аутоиммунная, невоспалительная тромботическая васкулопатия. Впервые был описан в 1986 г. английским ревматологом
- 10. Кожные проявления 40% Периферический тромбоз 64% Неврологические проявления 66% Поражение легких 20% Акушерская патология 80% Поражения
- 11. 3.Нейросифилис Проникновение бледных трепонем в клетки головного мозга с последующим их разрушением. Усиливается разрушение личности, ухудшается
- 12. 4.Криптококковая инфекция Заболевание начинается, если организм ослаб (снижение иммунитета, глубокий иммунодефицит, когда число CD меньше 50-100/мкл):
- 13. Головная боль (лоб, виски), головокружение, нарушения зрения, повышенная возбудимость, малые эпилептические припадки по джексоновскому типу (
- 14. Нейросифилис и криптококковый менингоэнцефалит Могут проявляться деменцией и миоклониями, нарастающими довольно быстро. Решающими в данном случае
- 15. 3.1. 5.Группа болезней, проявляющихся миоклонус-эпилепсией, мнестико-интеллектуальными нарушениями и атаксией может напоминать БКЯ, однако начало их в
- 16. 5.1.Митохондриальная энцефаломиопатия с синдромом «рваных» красных волокон (синдром MERRF) Наследуется по материнской линии Клиника: Низкорослость, снижение
- 17. 5.2. Болезнь Лафора Наследственная миоклоническая эпилепсия, при которой наблюдается отложения полисахаридных веществ в различных тканях (церебральных
- 18. 5.3. Болезнь Унферрихта- Лундборга (Семейная миоклония, миоклонус-эпилепсия, прогрессирующий миоклонус) Хроническое прогрессирующее наследственное заболевание ЦНС с аутосомно-рецессивным
- 19. Диагностика Клиника в виде тяжёлого стимул-сенситивного миоклонуса Возраст ЭЭГ (фотосенситивность, замедление фоновой ритмики, генерализованные высокоамплитудные полиспайки,
- 20. 5.4. Нейрональный цероидный липофусциноз (восковидные липофусцинозы нейронов) Нейродегенеративные наследственные заболевания, относящиеся к лизосомным болезням накопления (накопление
- 21. Клиника: Атрофия головного мозга Судороги (миоклонические) Диагностика: Электронная микроскопия (включения пигмента липофусцина в лизосомах тканях (печень,
- 22. 6. СПИД-деменция Начальные проявления СПИД-деменция также могут напоминать БКЯ по началу заболевания, клиническим признакам, отсутствию отклонений
- 23. Дифференциальных диагноз СГШШ проводится с: Оливопонтоцеребеллярной атаксией Гепатоцеребральной дегенерацией Рассеянным склерозом Семейной формой болезни Альцгеймера Метахроматической
- 24. 1. Оливопонтоцеребеллярная атаксия Наследственное заболевание, связанное с дефектом фермента дегидрогеназы глутамата в фибробластах и лейкоцитах. Глутаминовая
- 25. В норме глутамат – это нейромедиатор, который передает нервный импульс от оливы, ядер моста, спинного мозга
- 26. Деменция Нарушение психики и поведения Атаксия мозжечка Двигательные нарушения (экстрапирамидные расстройства, поражение черной субстанции и подкорковых
- 27. 2. Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация или болезнь Вильсона-Коновалова) Наследственный дефект метаболизма меди (20 хр), связанный с
- 28. 3. Болезнь Рефсума (полиневритоподобная гемератюпическая гередоатаксия) Редкое наследственное заболевание с накоплением фитановых кислот в центральной и
- 29. ПОСКОЛЬКУ БЫЛО ПОКАЗАНО, ЧТО ИНФЕКЦИОННЫЙ ПРИОННЫЙ БЕЛОК ОТЛИЧАЕТСЯ ОТ ОБЫЧНОГО ТРЕТИЧНОЙ СТРУКТУРОЙ, Т.Е. КОНФОРМАЦИОННО, ВСКОРЕ ПОЯВИЛОСЬ
- 31. Предполагаемое изменение характера укладки полипептидной цепи при превращении белка РгРC (а) в прион РгРSc (б)
- 32. Микрофотография конечной фазы губкообразных изменений в коре большого мозга:
- 33. Конформационные болезни человека
- 34. В 1854г. Р. Вирхов показал, что изменения эти связаны с появлением в органах особого вещества, которое
- 35. Амилоид – гликопротеид, основным компонентом которого являются фибриллярные белки ( F-компонент). Они образуют фибриллы, имеющие характерную
- 36. Molecular Mechanisms of Amyloidosis Giampaolo Merlini, M.D., and Vittorio Bellotti, M.D., Ph.D./ N Med 2003; 349:
- 37. 4. Болезнь Альцгеймера БА имеет достаточно сложный патогенез, но по процессу образования амилоидных включений данная патология
- 38. БА – медленно и длительно текущее заболевание, имеющее 3 основные стадии Образование и накопление Аβ –
- 39. Патогенез
- 41. По мнению D.Westaway (1998г) сходство симптоматики этих болезней отражает близость фундаментальных элементов патогенеза. Эти болезни могут
- 42. СПАСИБО ЗА ВНИМАНИЕ
- 44. Скачать презентацию
 Уход за центральным венозным катетером
Уход за центральным венозным катетером Дефицит массы тела как фактор риска заболеваний
Дефицит массы тела как фактор риска заболеваний Оценка эффективности использования аппарата Тонзиллор для лечения хронического тонзиллита
Оценка эффективности использования аппарата Тонзиллор для лечения хронического тонзиллита Бронхоспастический синдром
Бронхоспастический синдром Гипофизарный нанизм
Гипофизарный нанизм Вирус простого герпеса
Вирус простого герпеса Синтетикалық антимикробты заттар
Синтетикалық антимикробты заттар Клинико-лабораторная дифференциальная диагностика и интенсивная терапия приобретенных коагулопатий
Клинико-лабораторная дифференциальная диагностика и интенсивная терапия приобретенных коагулопатий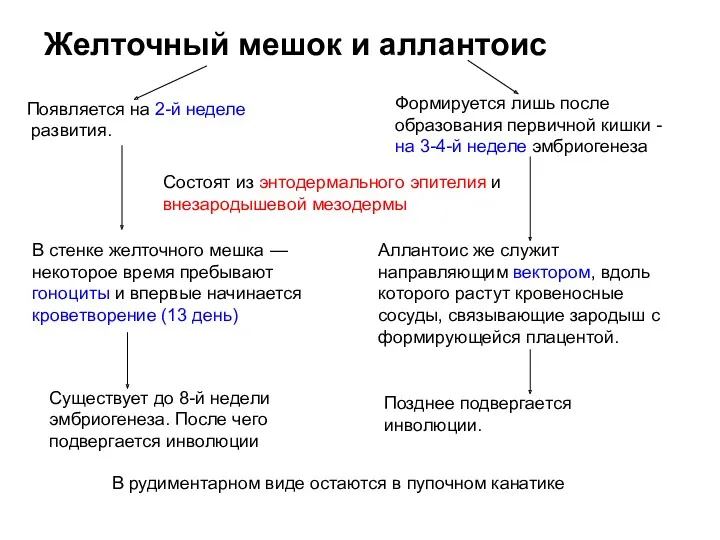 Желточный мешок и аллантоис
Желточный мешок и аллантоис Семиотика и диагностика урологических заболеваний. Кафедра урологии и андрологии ХГМУ
Семиотика и диагностика урологических заболеваний. Кафедра урологии и андрологии ХГМУ Ішкі аурулар пропедевтикасы
Ішкі аурулар пропедевтикасы Государственное регулирование ценообразования на лекарственные препараты
Государственное регулирование ценообразования на лекарственные препараты Лимфалық ісіктер БМСК жағдайында диагнозын анықтау
Лимфалық ісіктер БМСК жағдайында диагнозын анықтау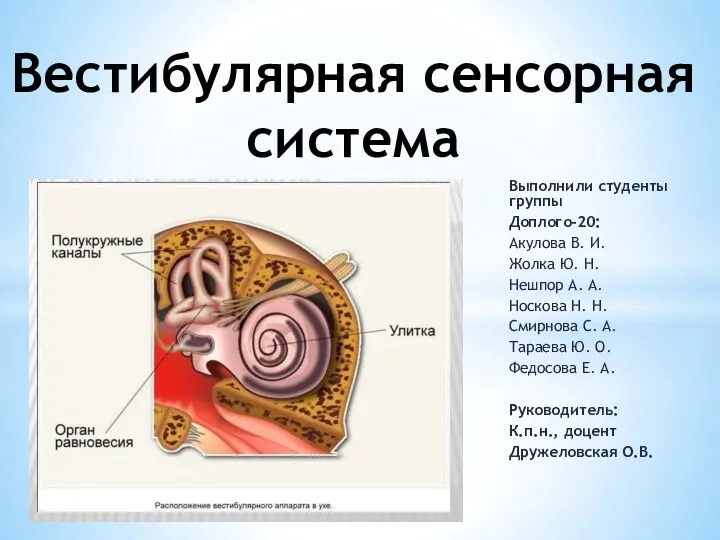 Вестибулярная сенсорная система
Вестибулярная сенсорная система Влияние алкоголя на здоровье человека
Влияние алкоголя на здоровье человека Острая травма кровеносных сосудов
Острая травма кровеносных сосудов Заболевания глаз. Рефракция и аккомодация
Заболевания глаз. Рефракция и аккомодация Хронический гепатит и цирроз печени
Хронический гепатит и цирроз печени Анемії у дітей. Епідеміологія анемій
Анемії у дітей. Епідеміологія анемій Сепсис. Этиологиясы. Патогенезі. Дагностикасы. Емделуі
Сепсис. Этиологиясы. Патогенезі. Дагностикасы. Емделуі История хирургии. Асптика. Антисептика. (лекция 1)
История хирургии. Асптика. Антисептика. (лекция 1) Актуальные проблемы педиатрии
Актуальные проблемы педиатрии Аномальные маточные кровотечения (АМК)
Аномальные маточные кровотечения (АМК) Синдром артериальной гипертензии. Гипертоническая болезнь
Синдром артериальной гипертензии. Гипертоническая болезнь Микобактерия туберкулеза. Микробиология
Микобактерия туберкулеза. Микробиология Доброкачественные и злокачественные опухоли женских половых органов
Доброкачественные и злокачественные опухоли женских половых органов Marfani sündroom
Marfani sündroom Когнитивно-поведенческая психотерапия в работе с пациентом с личностным расстройством
Когнитивно-поведенческая психотерапия в работе с пациентом с личностным расстройством