- Митохондриальная патология

Содержание
- 2. Митохондриальные болезни — группа патологических состояний, обусловленных клеточной энергетической недостаточностью изза нарушений биохимических процессов в митохондриях,
- 3. В настоящее время описано около 40 клинических форм МБ, для которых известны молекулярногенетический и биохимический дефект
- 4. Первые симптомы МБ носят неспецифический характер, что затрудняет их раннюю диагностику. У маленьких детей заболевание начинается
- 5. Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, Stroke-like episodes,митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды) - заболевание, обусловленное точечными
- 6. Возраст, в котором манифестирует заболевание, широко варьирует от младенческого до взрослого, однако чаще всего первые симптомы
- 7. Обследование включает проведение биохимических, морфологических и молекулярно-генетических исследований. Наиболее частая мутация - замена А на G
- 8. Синдром NARP (Neurogenic weakness, Ataxia, Retinitis Pigmentosa, синдром нейропатии, атаксии, пигментного ретинита) впервые описан в 1990
- 9. По данным лабораторных исследований нередко обнаруживают лактат-ацидоз, однако его может и не быть. При морфологическом исследовании
- 10. Синдром MERRF (Myoclonic Epilepsy with Ragged-Red Fibers, миоклонус-эпилепсия, «рваные» красные волокна) впервые описан в 1980 году.
- 11. Заболевание отличается выраженным клиническим полиморфизмом, включая семейный, и носит прогрессирующий характер. Возраст манифестации значительно варьирует от
- 12. Основные критерии синдрома MERRF: 1.митохондриальный тип наследования; 2.широкий возрастной диапазон манифестации болезни (3-65 лет); 3.сочетание симптомов
- 13. Синдром мионеврогастроинтестинальной невропатии ( MNGIE) . Заболевание обусловлено мутациями гена ТР, кодирующего тимидин фосфорилазу (TYMP; MIM
- 14. При ЭНМГ выявляют признаки аксональной демиелинизирующей полиневропатии. У большинства пациентов в цереброспинальной жидкости обнаруживают плеоцитоз. При
- 15. Синдром Вольфрама (синдром DIDMOAD - Diabetes Insipidus, Diabetes Mettitus, Optic Atrophy, Deafness, OMIM 598500) описан впервые
- 16. Симптомы синдрома Вольфрама. Заболевание развивается в раннем детском возрасте (1-8 лет). Начинается оно с появления симптомов
- 17. Основной составляющей лечебной тактики пациентов с данный прогрессирующим заболеванием является поддержка семей и обучение детей практическим
- 18. Синдром Кернса-Сейра - это заболевание впервые описано в 1958 г. Большинство случаев обусловлено крупными делениями мтДНК.
- 19. Заболевание манифестирует в возрасте 4-20 лет и включает триаду симптомов: офтальмоплегию с птозом верхнего века и
- 20. Синдром Пирсона-Марроу. Это заболевание, которое совсем не поддаётся лечению, начинает проявляться уже в первые недели после
- 21. Большинство больных погибают в первые 2 года жизни. Однако у тех лиц, которые выжили благодаря частым
- 22. До настоящего времени эффективное лечение митохондриальных болезней остаётся нерешённой проблемой. Это связано с несколькими факторами: трудностями
- 24. Скачать презентацию

Митохондриальные болезни — группа патологических состояний, обусловленных клеточной энергетической недостаточностью изза
Митохондриальные болезни — группа патологических состояний, обусловленных клеточной энергетической недостаточностью изза

В настоящее время описано около 40 клинических форм МБ, для которых
В настоящее время описано около 40 клинических форм МБ, для которых

Первые симптомы МБ носят неспецифический характер, что затрудняет их раннюю диагностику.
Первые симптомы МБ носят неспецифический характер, что затрудняет их раннюю диагностику.

Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, Stroke-like episodes,митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды)
Синдром MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, Stroke-like episodes,митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды)
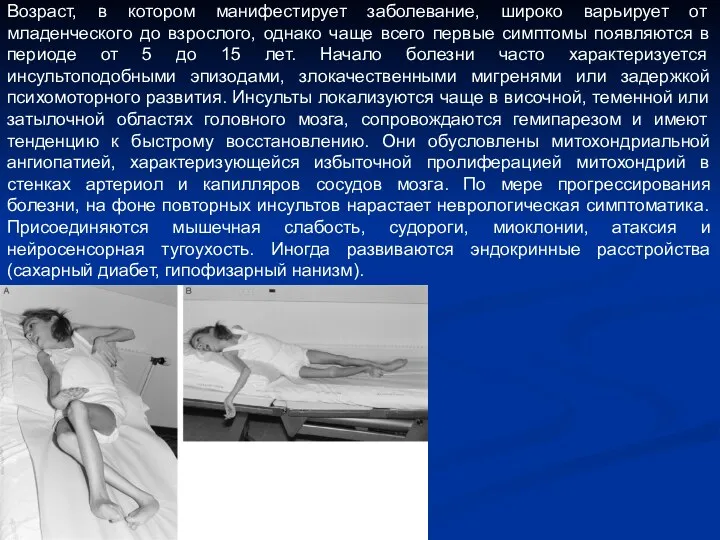
Возраст, в котором манифестирует заболевание, широко варьирует от младенческого до взрослого,
Возраст, в котором манифестирует заболевание, широко варьирует от младенческого до взрослого,

Обследование включает проведение биохимических, морфологических и молекулярно-генетических исследований. Наиболее частая мутация
Обследование включает проведение биохимических, морфологических и молекулярно-генетических исследований. Наиболее частая мутация
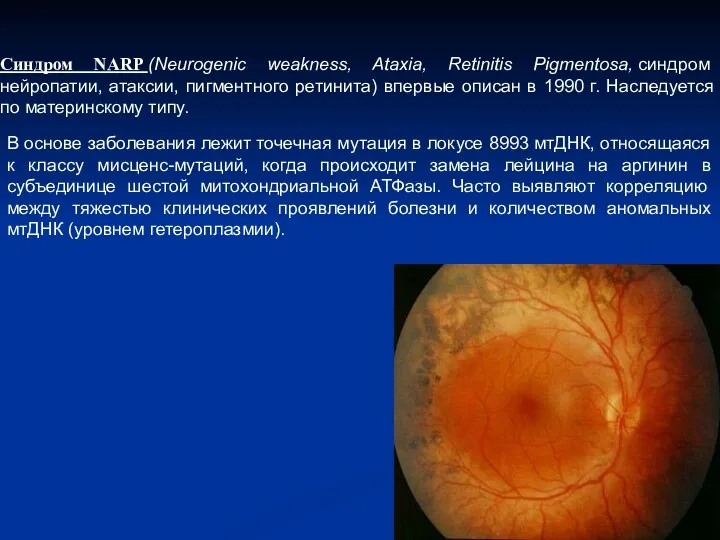
Синдром NARP (Neurogenic weakness, Ataxia, Retinitis Pigmentosa, синдром нейропатии, атаксии, пигментного ретинита) впервые
Синдром NARP (Neurogenic weakness, Ataxia, Retinitis Pigmentosa, синдром нейропатии, атаксии, пигментного ретинита) впервые

По данным лабораторных исследований нередко обнаруживают лактат-ацидоз, однако его может и
По данным лабораторных исследований нередко обнаруживают лактат-ацидоз, однако его может и
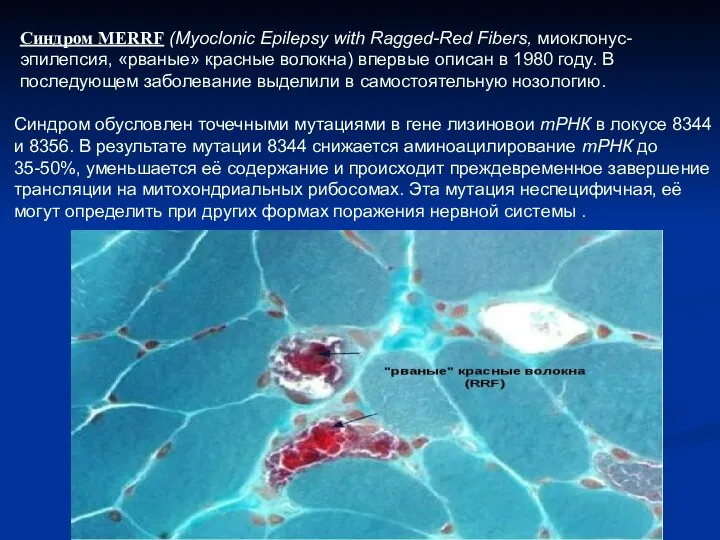
Синдром MERRF (Myoclonic Epilepsy with Ragged-Red Fibers, миоклонус-эпилепсия, «рваные» красные волокна) впервые описан
Синдром MERRF (Myoclonic Epilepsy with Ragged-Red Fibers, миоклонус-эпилепсия, «рваные» красные волокна) впервые описан

Заболевание отличается выраженным клиническим полиморфизмом, включая семейный, и носит прогрессирующий характер.
Заболевание отличается выраженным клиническим полиморфизмом, включая семейный, и носит прогрессирующий характер.

Основные критерии синдрома MERRF:
1.митохондриальный тип наследования;
2.широкий возрастной диапазон манифестации болезни (3-65
Основные критерии синдрома MERRF:
1.митохондриальный тип наследования;
2.широкий возрастной диапазон манифестации болезни (3-65

Синдром мионеврогастроинтестинальной невропатии ( MNGIE) .
Заболевание обусловлено мутациями гена ТР, кодирующего
Синдром мионеврогастроинтестинальной невропатии ( MNGIE) .
Заболевание обусловлено мутациями гена ТР, кодирующего

При ЭНМГ выявляют признаки аксональной демиелинизирующей полиневропатии. У большинства пациентов в
При ЭНМГ выявляют признаки аксональной демиелинизирующей полиневропатии. У большинства пациентов в

Синдром Вольфрама (синдром DIDMOAD - Diabetes Insipidus, Diabetes Mettitus, Optic Atrophy, Deafness, OMIM
Синдром Вольфрама (синдром DIDMOAD - Diabetes Insipidus, Diabetes Mettitus, Optic Atrophy, Deafness, OMIM

Симптомы синдрома Вольфрама. Заболевание развивается в раннем детском возрасте (1-8 лет).
Симптомы синдрома Вольфрама. Заболевание развивается в раннем детском возрасте (1-8 лет).

Основной составляющей лечебной тактики пациентов с данный прогрессирующим заболеванием является поддержка
Основной составляющей лечебной тактики пациентов с данный прогрессирующим заболеванием является поддержка
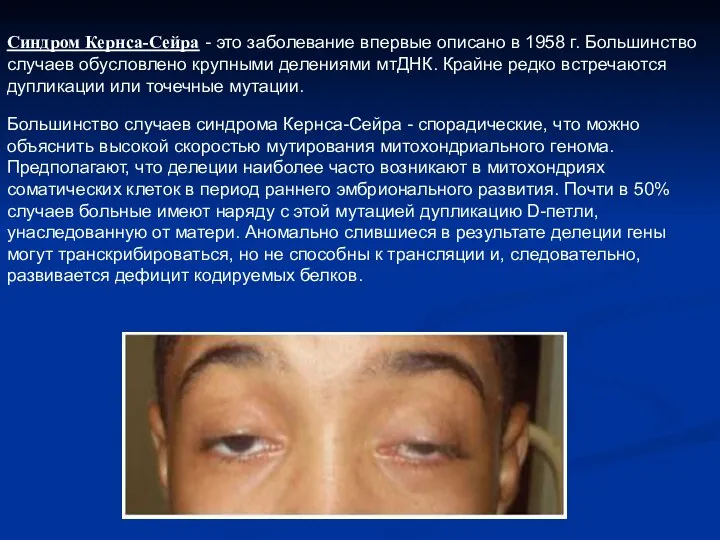
Синдром Кернса-Сейра - это заболевание впервые описано в 1958 г. Большинство
Синдром Кернса-Сейра - это заболевание впервые описано в 1958 г. Большинство

Заболевание манифестирует в возрасте 4-20 лет и включает триаду симптомов:
офтальмоплегию с
Заболевание манифестирует в возрасте 4-20 лет и включает триаду симптомов:
офтальмоплегию с

Синдром Пирсона-Марроу.
Это заболевание, которое совсем не поддаётся лечению, начинает проявляться уже
Синдром Пирсона-Марроу.
Это заболевание, которое совсем не поддаётся лечению, начинает проявляться уже
Большинство больных погибают в первые 2 года жизни. Однако у тех лиц, которые
Большинство больных погибают в первые 2 года жизни. Однако у тех лиц, которые
До настоящего времени эффективное лечение митохондриальных болезней остаётся нерешённой проблемой. Это
До настоящего времени эффективное лечение митохондриальных болезней остаётся нерешённой проблемой. Это
 Климат и микроклимат, гигиена организаций здравоохранения. (Лекция 6)
Климат и микроклимат, гигиена организаций здравоохранения. (Лекция 6) Диспансеризация беременных женщин. Ведение беременности у женщин групп риска. Медико-генетическое консультирование
Диспансеризация беременных женщин. Ведение беременности у женщин групп риска. Медико-генетическое консультирование Исключение острой хирургической патологии при остром животе у женщин
Исключение острой хирургической патологии при остром животе у женщин Опыт лечения опухолей опорно-двигательной системы
Опыт лечения опухолей опорно-двигательной системы Рекуррентные ОРВИ в практике педиатра
Рекуррентные ОРВИ в практике педиатра Реабилитация при невритах, невралгиях
Реабилитация при невритах, невралгиях Хронический холецистит. Желчнокаменная болезнь
Хронический холецистит. Желчнокаменная болезнь Inflammatory Bowel Diseases
Inflammatory Bowel Diseases Электрокардиостимуляция
Электрокардиостимуляция Наследственные атаксии Пьера-Мари, Фридрейха
Наследственные атаксии Пьера-Мари, Фридрейха Этапы организации Мониторинга факторов риска. Формирование выборки. Подготовительный этап. Приглашение на обследование
Этапы организации Мониторинга факторов риска. Формирование выборки. Подготовительный этап. Приглашение на обследование Предмет и задачи медицинской микробиологии
Предмет и задачи медицинской микробиологии Гормондар, жіктелуі, түрлері, физиологиялық рөлдері орындағандар
Гормондар, жіктелуі, түрлері, физиологиялық рөлдері орындағандар Острая ревматическая лихорадка у детей
Острая ревматическая лихорадка у детей Акушерская техника
Акушерская техника Көпіршікті дерматоздар: Ұшық тәрізді Дюринг дерматозы
Көпіршікті дерматоздар: Ұшық тәрізді Дюринг дерматозы Қанжасау. Айырша без
Қанжасау. Айырша без Подагра. Стадии развития подагры
Подагра. Стадии развития подагры Кровотечения в последовом и послеродовом периодах
Кровотечения в последовом и послеродовом периодах Лабораторная диагностика синдрома системной воспалительной реакции и сепсиса
Лабораторная диагностика синдрома системной воспалительной реакции и сепсиса Регенерация костной ткани
Регенерация костной ткани О хирургических методах лечения ожирения и метаболических нарушений
О хирургических методах лечения ожирения и метаболических нарушений Психотерапия в узком смысле. Групповая и индивидуальная формы психотерапии
Психотерапия в узком смысле. Групповая и индивидуальная формы психотерапии Плацентарная недостаточность. Гипоксия плода. Синдром задержки роста плода
Плацентарная недостаточность. Гипоксия плода. Синдром задержки роста плода Философия сестринского дела
Философия сестринского дела Физиология родов (причины наступления родов, регуляция родовой деятельности). Периоды родов. Адаптация плода к родам
Физиология родов (причины наступления родов, регуляция родовой деятельности). Периоды родов. Адаптация плода к родам Венозный доступ в интенсивной терапии
Венозный доступ в интенсивной терапии Иммунная система и клеточные взаимодействия в иммунных реакциях
Иммунная система и клеточные взаимодействия в иммунных реакциях