- Наследственные атаксии Пьера-Мари, Фридрейха

Содержание
- 2. ПЛАН: I.ВВЕДЕНИЕ. ОПРЕДЕЛЕНИЕ II.ОСНОВНАЯ ЧАСТЬ КЛИНИКА ДИАГНОСТИКА ЛЕЧЕБНАЯ ТАКТИКА ПРОГНОЗ III.ЗАКЛЮЧЕНИЯ IV. СПИСОК ЛИТЕРАТУР.
- 3. ЦЕЛЬ: Дать общее понятие и лечении этой болезни , также сопровождающиеся развитие осложнений после лечения .
- 4. ОПРЕДЕЛЕНИЕ Атаксия (греч. ataxia — беспорядок, «а» — отрицательная частица и «taxis» — порядок) Атаксия —
- 5. Болезнь Фридрейха Атаксия Dридрейха – наследственное нейродегенеративное заболевание, начинающееся в детском или юношеском возрасте и характеризующееся
- 7. Первые симптомы заболевания возникают обычно на 1—2-м десятилетии жизни, чаще всего в препубертатном периоде, характеризуются незаметным
- 8. Заболевание обычно манифестирует появлением неловкости, неуверенности при ходьбе, особенно в темноте (признак заднестолбовой атаксии), больные начинают
- 9. Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько лет опережать манифестацию других симптомов
- 10. Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной) чувствительности. Довольно рано у больных
- 11. ЭКСТРАНЕВРАЛЬНЫЕ СИМПТОМЫ
- 12. Основными критериями диагноза болезни Фридрейха являются: 1. аутосомно-рецессивный тип наследования; 2. дебют в подростковом, реже в
- 13. А — Сибсы с семенной атаксиеи Фридрейха Б — Схема поражения ' при болезни Фпидреиха, семейной
- 14. ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ. Заболевание распознается на основании характерных симптомов - деформаций стоп по типу стопы
- 15. Дифференцировать заболевание следует от атаксии с изолированным дефицитом витамина Е (мутация в хромосоме 8q), рассеянного склероза,
- 16. ЛЕЧЕНИЕ. Применяют симптоматические средства: общеукрепляющие препараты (витамины, аминокислоты, ноотропные 492 и др.), лечебная физкультура, массаж. В
- 17. ПРОГНОЗ. Кардиомиопатия приводит к застойной сердечной недостаточности. Продолжительность жизни больных редко превышает 30 лет.
- 18. НАСЛЕДСТВЕННАЯ МОЗЖЕЧКОВАЯ АТАКСИЯ ПЬЕРА МАРИ Мозжечковая атаксия Пьера Мари - наследственное дегенеративное заболевание с преимущественным поражением
- 19. КЛИНИЧЕСКАЯ КАРТИНА. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые
- 20. ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии
- 21. ЛЕЧЕНИЕ Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном это антидепрессанты (амитриптилин, флуоксетин,
- 22. Заключение Известно, что в последние десятилетия появилось большое количество мутагенных факторов: радиация, химические мутагены (лекарственные препараты,
- 23. Список литератур Гусев Е.И. Неврология. Национальное руководство. Краткое издание. ГЭОТАР-Медиа, 2014.- 688с. Скоромец А.А., Скоромец А.П.,
- 25. Скачать презентацию

ПЛАН:
I.ВВЕДЕНИЕ.
ОПРЕДЕЛЕНИЕ
II.ОСНОВНАЯ ЧАСТЬ
КЛИНИКА
ДИАГНОСТИКА ЛЕЧЕБНАЯ ТАКТИКА
ПРОГНОЗ
III.ЗАКЛЮЧЕНИЯ
IV. СПИСОК ЛИТЕРАТУР.
ПЛАН:
I.ВВЕДЕНИЕ.
ОПРЕДЕЛЕНИЕ
II.ОСНОВНАЯ ЧАСТЬ
КЛИНИКА
ДИАГНОСТИКА ЛЕЧЕБНАЯ ТАКТИКА
ПРОГНОЗ
III.ЗАКЛЮЧЕНИЯ
IV. СПИСОК ЛИТЕРАТУР.

ЦЕЛЬ:
Дать общее понятие
и лечении этой болезни ,
ЦЕЛЬ:
Дать общее понятие
и лечении этой болезни ,
ОПРЕДЕЛЕНИЕ
Атаксия (греч. ataxia — беспорядок,
«а» — отрицательная частица и
ОПРЕДЕЛЕНИЕ
Атаксия (греч. ataxia — беспорядок,
«а» — отрицательная частица и
Болезнь Фридрейха Атаксия Dридрейха – наследственное нейродегенеративное заболевание, начинающееся в детском
Болезнь Фридрейха Атаксия Dридрейха – наследственное нейродегенеративное заболевание, начинающееся в детском
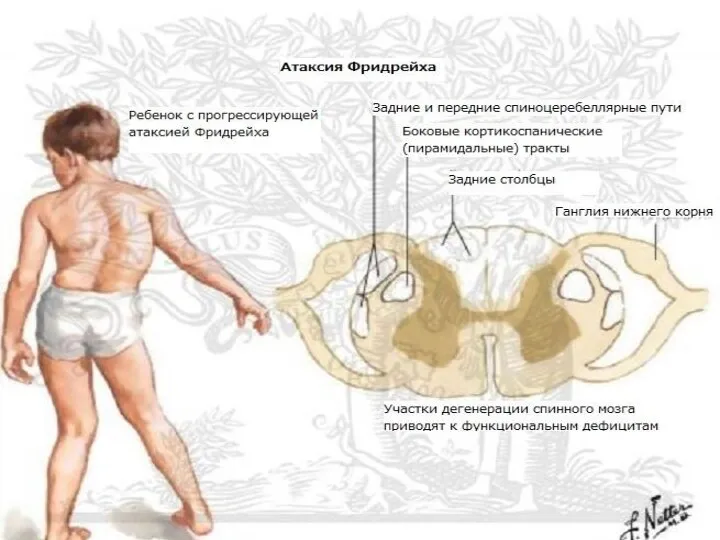

Первые симптомы заболевания возникают обычно на 1—2-м десятилетии жизни, чаще всего
Первые симптомы заболевания возникают обычно на 1—2-м десятилетии жизни, чаще всего
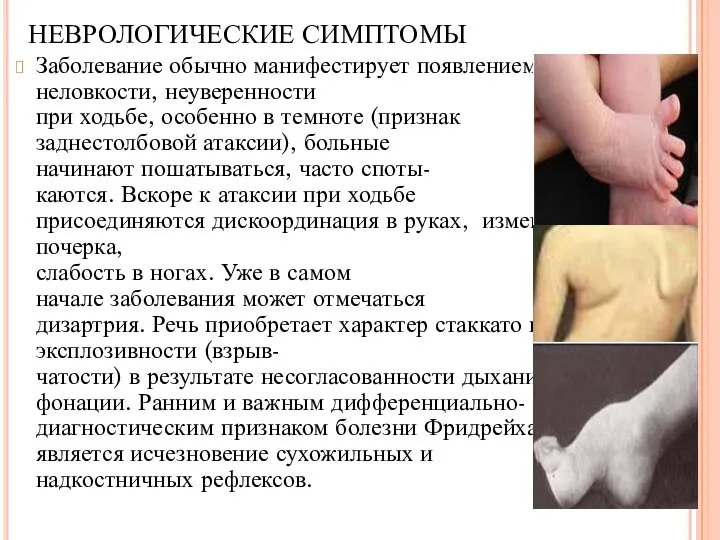
Заболевание обычно манифестирует появлением неловкости, неуверенности
при ходьбе, особенно в темноте
Заболевание обычно манифестирует появлением неловкости, неуверенности при ходьбе, особенно в темноте

Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько
Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько

Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной)
Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной)

ЭКСТРАНЕВРАЛЬНЫЕ СИМПТОМЫ
ЭКСТРАНЕВРАЛЬНЫЕ СИМПТОМЫ

Основными критериями диагноза болезни Фридрейха являются:
1. аутосомно-рецессивный тип наследования;
2. дебют в
Основными критериями диагноза болезни Фридрейха являются:
1. аутосомно-рецессивный тип наследования;
2. дебют в
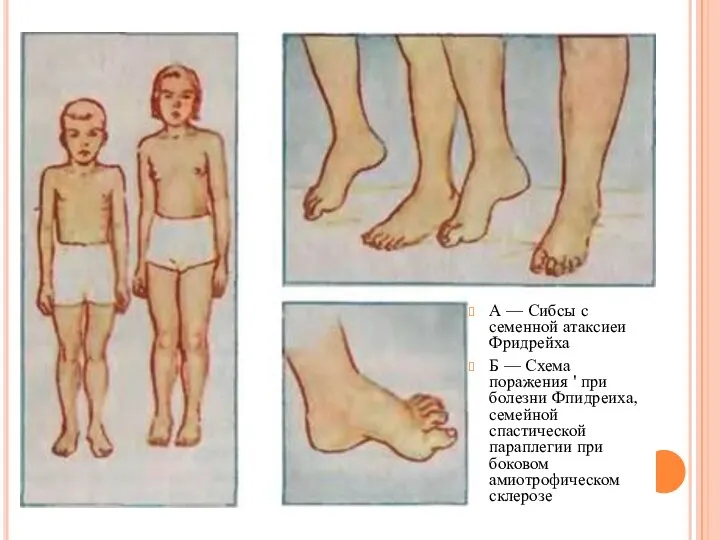
А — Сибсы с семенной атаксиеи Фридрейха
Б — Схема поражения '
А — Сибсы с семенной атаксиеи Фридрейха
Б — Схема поражения '

ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ.
Заболевание распознается на основании характерных симптомов - деформаций
ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ.
Заболевание распознается на основании характерных симптомов - деформаций
Дифференцировать заболевание следует от атаксии с изолированным дефицитом витамина Е (мутация
Дифференцировать заболевание следует от атаксии с изолированным дефицитом витамина Е (мутация
ЛЕЧЕНИЕ.
Применяют симптоматические средства: общеукрепляющие препараты (витамины, аминокислоты, ноотропные 492 и др.),
ЛЕЧЕНИЕ.
Применяют симптоматические средства: общеукрепляющие препараты (витамины, аминокислоты, ноотропные 492 и др.),

ПРОГНОЗ.
Кардиомиопатия приводит к застойной сердечной недостаточности. Продолжительность жизни больных редко превышает
ПРОГНОЗ.
Кардиомиопатия приводит к застойной сердечной недостаточности. Продолжительность жизни больных редко превышает

НАСЛЕДСТВЕННАЯ МОЗЖЕЧКОВАЯ АТАКСИЯ ПЬЕРА МАРИ
Мозжечковая атаксия Пьера Мари - наследственное дегенеративное
НАСЛЕДСТВЕННАЯ МОЗЖЕЧКОВАЯ АТАКСИЯ ПЬЕРА МАРИ
Мозжечковая атаксия Пьера Мари - наследственное дегенеративное

КЛИНИЧЕСКАЯ КАРТИНА.
Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь,
КЛИНИЧЕСКАЯ КАРТИНА.
Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь,

ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ.
Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии
ДИАГНОЗ И ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ.
Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии

ЛЕЧЕНИЕ
Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном
ЛЕЧЕНИЕ
Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном

Заключение
Известно, что в последние десятилетия появилось большое количество мутагенных факторов: радиация,
Заключение
Известно, что в последние десятилетия появилось большое количество мутагенных факторов: радиация,

Список литератур
Гусев Е.И. Неврология. Национальное руководство. Краткое издание. ГЭОТАР-Медиа, 2014.- 688с.
Скоромец
Список литератур
Гусев Е.И. Неврология. Национальное руководство. Краткое издание. ГЭОТАР-Медиа, 2014.- 688с.
Скоромец
 Стоматологиядағы кірісулер кезіндегі шұғыл көмек
Стоматологиядағы кірісулер кезіндегі шұғыл көмек Половое воспитание. Инфекции, передаваемые половым путём. 9 класс
Половое воспитание. Инфекции, передаваемые половым путём. 9 класс Көпіршікті дерматоздар (пемфигус)
Көпіршікті дерматоздар (пемфигус) Этапы обработки медицинских изделий. Тема 5
Этапы обработки медицинских изделий. Тема 5 Гиперкинетический синдром у детей
Гиперкинетический синдром у детей Бауме бойынша тістесу түрлері
Бауме бойынша тістесу түрлері Жас стоматолог
Жас стоматолог Шизофрения. Признаки шизофрении
Шизофрения. Признаки шизофрении Полуколичественный метод определения ДНК онкогенных типов ВПЧ-Digene-тест, онкоцитологическое исследование ASC-US
Полуколичественный метод определения ДНК онкогенных типов ВПЧ-Digene-тест, онкоцитологическое исследование ASC-US Surgical revascularization of myocardium
Surgical revascularization of myocardium Haemolytic disease of the fetus and newborn. Rh isoimmunization
Haemolytic disease of the fetus and newborn. Rh isoimmunization Рак щитовидной железы
Рак щитовидной железы Відмороження. Визначення поняття “відмороження”
Відмороження. Визначення поняття “відмороження” Артерия гистологиясы
Артерия гистологиясы Трансплантация почки
Трансплантация почки Теория медицинского диагноза и клинико-анатомический анализ летальных исходов. Теория и практика
Теория медицинского диагноза и клинико-анатомический анализ летальных исходов. Теория и практика Современные проблемы диагностики сепсиса
Современные проблемы диагностики сепсиса Искусственное кровообращение
Искусственное кровообращение Острый тонзиллит
Острый тонзиллит Кариес зуба
Кариес зуба Ноотропные препараты
Ноотропные препараты Первая помощь при ожогах и обморожениях
Первая помощь при ожогах и обморожениях Основные приемы классического массажа
Основные приемы классического массажа Зәр шығару жүйесінің сәулелік диагностика әдістері
Зәр шығару жүйесінің сәулелік диагностика әдістері Зерттеу сұрағы мен ақпаратты іздеу
Зерттеу сұрағы мен ақпаратты іздеу Ламбдацизм и методы его исправления
Ламбдацизм и методы его исправления Определение содержания гемоглобина по методу Сали. Лабораторная работа № 3
Определение содержания гемоглобина по методу Сали. Лабораторная работа № 3 Хроническая лучевая болезнь
Хроническая лучевая болезнь