- Мукополисахаридоз

Содержание
- 2. Содержание Определение заболевания Классификация заболевания Механизм развития синдрома Гурлера Фенотип больного синдромом Гурлера Клиническое проявление Диагностика
- 3. Определение заболевания Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ),
- 4. Классификация заболевания
- 5. Кодирование по МКБ-10 E76.0 - Мукополисахаридоз I типа Согласно клиническим проявлениям выделяют 3 формы мукополисахаридоза I
- 6. Механизм развития: Наследуется по аутосомно-рецессивному типу Мутация гена IDUA Дефицит фермента альфа-L-идуронидазы Накопление субстрата при недостаточной
- 7. Фенотип больного синдромом Гурлера
- 8. Клиническое проявление Характерным симптомом для неё является деформация черепа с развитием грубых черт лица. К двум
- 9. Диагностика Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение зрения; помутнение
- 10. Лечение Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ). ФЗТ проводится ларонидазой (код ATX A16AB05); коррекцию сердечно-сосудистой
- 11. Мукополисахаридоз II типа Мукополисахаридоз II типа наследственная лизосомная болезнь накопления, с Хсцепленным рецессивным типом наследования, которая
- 12. Механизм развития: Наследуется с Х-сцепленному рецессивному типу Мутация гена IDS Снижение активности идуронат-2-сульфатазы(I2S) Аккумуляция гликозаминогликанов (ГАГ)
- 13. Фенотип больного синдромом Хантера
- 14. Клиническое проявление Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и
- 15. Диагностика Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение зрения; помутнение
- 16. Лечение Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ) препаратом идурсульфаза (код АТХ A16AB09); коррекцию сердечно-сосудистой недостаточности,
- 17. Мукополисахаридоз III типа Мукополисахаридоз III типа (Синдром Cанфилиппо) - наследственная лизосомальная болезнь накопления, генетически гетерогенная, обусловленная
- 18. Механизм развития: Наследуется по аутосомно-рецессивному типу Мутация гена SGSH Нарушение кодирование гепаран-N-сульфатазу Локализации в хромосомной области
- 19. Фенотип больного синдромом Санфилиппо
- 20. Клиническое проявление В раннем периоде: ребенок начинает отставать в развитии от своих сверстников; ребенок не отличается
- 21. Диагностика Жалобы и анамнез: нарушение поведения; гиперактивность; нарушение сна; нарушение глотания; судороги; задержка речевого развития; снижение
- 22. Лечение Консервативное лечение: лечение поведенческих нарушений проводится психоневрологом, рекомендовано использование седативные средства, транквилизаторы, корректоры поведения; при
- 24. Скачать презентацию

Содержание
Определение заболевания
Классификация заболевания
Механизм развития синдрома Гурлера
Фенотип больного синдромом Гурлера
Клиническое проявление
Диагностика
Лечение
Мукополисахаридоза
Содержание
Определение заболевания
Классификация заболевания
Механизм развития синдрома Гурлера
Фенотип больного синдромом Гурлера
Клиническое проявление
Диагностика
Лечение
Мукополисахаридоза

Определение заболевания
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с
Определение заболевания
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с

Классификация заболевания
Классификация заболевания

Кодирование по МКБ-10
E76.0 - Мукополисахаридоз I типа
Согласно клиническим проявлениям выделяют 3
Кодирование по МКБ-10
E76.0 - Мукополисахаридоз I типа
Согласно клиническим проявлениям выделяют 3
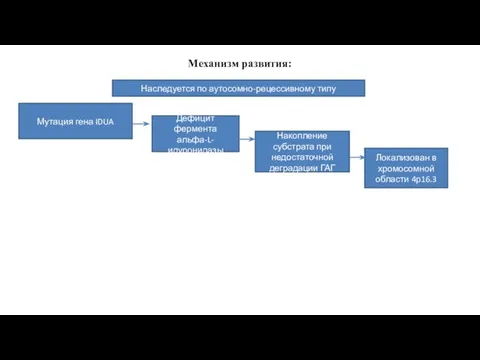
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена IDUA
Дефицит фермента альфа-L-идуронидазы
Накопление субстрата при
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена IDUA
Дефицит фермента альфа-L-идуронидазы
Накопление субстрата при

Фенотип больного синдромом Гурлера
Фенотип больного синдромом Гурлера

Клиническое проявление
Характерным симптомом для неё является деформация черепа с развитием грубых
Клиническое проявление
Характерным симптомом для неё является деформация черепа с развитием грубых

Диагностика
Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение
Диагностика
Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение

Лечение
Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ). ФЗТ проводится ларонидазой (код ATX
Лечение
Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ). ФЗТ проводится ларонидазой (код ATX

Мукополисахаридоз II типа
Мукополисахаридоз II типа наследственная лизосомная болезнь накопления, с Хсцепленным
Мукополисахаридоз II типа
Мукополисахаридоз II типа наследственная лизосомная болезнь накопления, с Хсцепленным
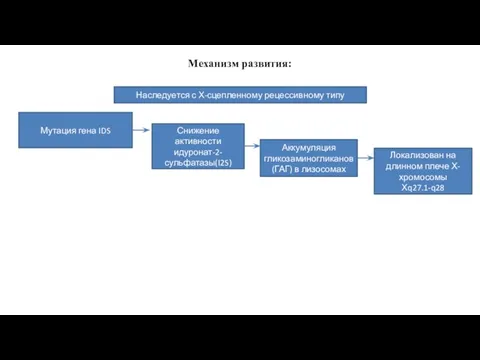
Механизм развития:
Наследуется с Х-сцепленному рецессивному типу
Мутация гена IDS
Снижение активности идуронат-2-сульфатазы(I2S)
Аккумуляция гликозаминогликанов
Механизм развития:
Наследуется с Х-сцепленному рецессивному типу
Мутация гена IDS
Снижение активности идуронат-2-сульфатазы(I2S)
Аккумуляция гликозаминогликанов

Фенотип больного синдромом Хантера
Фенотип больного синдромом Хантера

Клиническое проявление
Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт
Клиническое проявление
Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт

Диагностика
Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение
Диагностика
Жалобы и анамнез: огрубение черт лица; частые респираторные заболевания; грыжи; снижение слуха; снижение

Лечение
Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ) препаратом идурсульфаза (код АТХ A16AB09); коррекцию
Лечение
Консервативное лечение: проведение ферментной заместительной терапии (ФЗТ) препаратом идурсульфаза (код АТХ A16AB09); коррекцию

Мукополисахаридоз III типа
Мукополисахаридоз III типа (Синдром Cанфилиппо) - наследственная лизосомальная болезнь накопления,
Мукополисахаридоз III типа
Мукополисахаридоз III типа (Синдром Cанфилиппо) - наследственная лизосомальная болезнь накопления,

Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена SGSH
Нарушение кодирование гепаран-N-сульфатазу
Локализации в хромосомной
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена SGSH
Нарушение кодирование гепаран-N-сульфатазу
Локализации в хромосомной

Фенотип больного синдромом Санфилиппо
Фенотип больного синдромом Санфилиппо

Клиническое проявление
В раннем периоде: ребенок начинает отставать в развитии от своих сверстников; ребенок
Клиническое проявление
В раннем периоде: ребенок начинает отставать в развитии от своих сверстников; ребенок

Диагностика
Жалобы и анамнез: нарушение поведения; гиперактивность; нарушение сна; нарушение глотания; судороги; задержка речевого развития; снижение слуха; огрубение
Диагностика
Жалобы и анамнез: нарушение поведения; гиперактивность; нарушение сна; нарушение глотания; судороги; задержка речевого развития; снижение слуха; огрубение

Лечение
Консервативное лечение: лечение поведенческих нарушений проводится психоневрологом, рекомендовано использование седативные средства,
Лечение
Консервативное лечение: лечение поведенческих нарушений проводится психоневрологом, рекомендовано использование седативные средства,
 Экстрагенитальные заболевания, вызывающие клинику острого живота
Экстрагенитальные заболевания, вызывающие клинику острого живота Подход к пациенту с политравмой
Подход к пациенту с политравмой Врожденный и приобретенный иммунитет. Клеточные и гуморальные механизмы
Врожденный и приобретенный иммунитет. Клеточные и гуморальные механизмы Antigeny
Antigeny Маңызды класс аурулары деп. Туберкулез
Маңызды класс аурулары деп. Туберкулез Боковой амиотрофический склероз
Боковой амиотрофический склероз Гигиенические требования к рентгенологическим и радиологическим отделениям больниц
Гигиенические требования к рентгенологическим и радиологическим отделениям больниц Искусственная инсеминация
Искусственная инсеминация Синдром серцевої та судинної недостатності при захворюваннях серцево–судинної системи
Синдром серцевої та судинної недостатності при захворюваннях серцево–судинної системи Патофизиология ожоговой болезни. Интенсивная терапия ожоговой болезни и ожогового шока у детей
Патофизиология ожоговой болезни. Интенсивная терапия ожоговой болезни и ожогового шока у детей Көмекей, кеңірдек, бронхылар. Өкпенің құрылымы мен функциялары. Плевра қойнаулары
Көмекей, кеңірдек, бронхылар. Өкпенің құрылымы мен функциялары. Плевра қойнаулары Профессиональные компетенции медсестры при заболеваниях органов дыхания у детей
Профессиональные компетенции медсестры при заболеваниях органов дыхания у детей Модуль 2. Оказание первой помощи при отсутствии сознания, остановке дыхания и кровообращения
Модуль 2. Оказание первой помощи при отсутствии сознания, остановке дыхания и кровообращения Диагностика ишемической болезни сердца
Диагностика ишемической болезни сердца Подагра
Подагра Боль. Местная анестезия, блокады и общее обезболивание
Боль. Местная анестезия, блокады и общее обезболивание Рак пищевода
Рак пищевода Просветление. Рентгенопульмонология
Просветление. Рентгенопульмонология Нові підходи до лікування гострої респіраторної вірусної інфекції у дітей
Нові підходи до лікування гострої респіраторної вірусної інфекції у дітей Система комп’ютерного моделювання процесів життєдіяльності органів та систем організму СКІФ
Система комп’ютерного моделювання процесів життєдіяльності органів та систем організму СКІФ Жедел холецистит. Жіктемесі, диагностикасы, саралау диагностикасы. Аурудың асқынулары,асқынуларды диагностикалау
Жедел холецистит. Жіктемесі, диагностикасы, саралау диагностикасы. Аурудың асқынулары,асқынуларды диагностикалау Первая помощь при ранениях и кровотечениях. СОЦМК
Первая помощь при ранениях и кровотечениях. СОЦМК Наследственные атаксии Пьера-Мари, Фридрейха
Наследственные атаксии Пьера-Мари, Фридрейха 20231031_zhiznennyy_tsikl_virusov
20231031_zhiznennyy_tsikl_virusov Тірек-қимыл жүйесінің аурулары
Тірек-қимыл жүйесінің аурулары Галактоземия
Галактоземия Қан тобын анықтау
Қан тобын анықтау Азбука СПИДа
Азбука СПИДа