- Мукополисахаридоз типа I-Н (синдром Гурлер)

Содержание
- 2. Мукополисахаридоз типа I-Н (синдром Гурлер) - - наследственные заболевания обмена веществ, относящиеся к группе лизосомных болезней
- 3. Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер): - аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене лизосомного
- 4. Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера) Патоморфологическая картина: наблюдается утолщение костей черепа
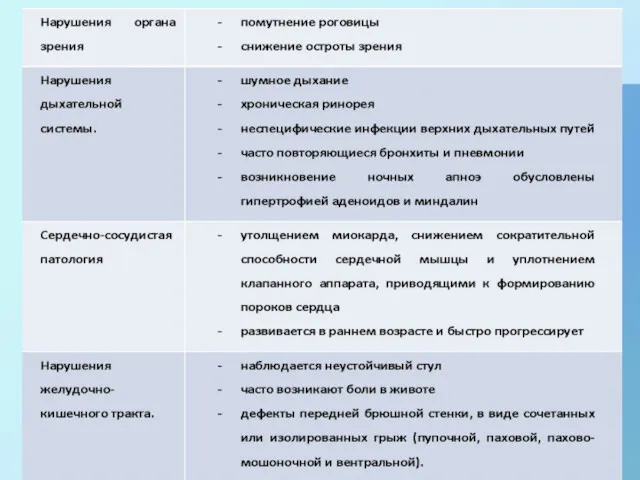
- 5. Нарушения при МПС I-H:
- 8. Особенности фенотипа. Характерны изменения черт лица по типу «гаргоилизма», которые становятся очевидными к концу первого года
- 9. Клиническая картина МПС I-H: Первые клинические признаки заболевания появляются на первом году жизни. В ряде случаев,
- 10. Клиническая диагностика В ряде случаев, выраженный клинический полиморфизм МПС I приводит к ошибочной диагностике, что в
- 11. Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера): Подтверждающая биохимическая диагностика МПС I заключается в определении уровня экскреции
- 12. Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера): Заменительная терапия. Трансплантация стволовых клеток. Хирургическая коррекция глаукомы, скелетных аномалий,
- 14. Скачать презентацию

Мукополисахаридоз типа I-Н (синдром Гурлер) -
- наследственные заболевания обмена веществ,
Мукополисахаридоз типа I-Н (синдром Гурлер) -
- наследственные заболевания обмена веществ,

Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер):
- аутосомно-рецессивное заболевание, обусловленное мутациями
Что провоцирует Мукополисахаридоз типа I-Н (синдром Гурлер):
- аутосомно-рецессивное заболевание, обусловленное мутациями

Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера)
Патоморфологическая картина:
наблюдается
Патогенез (что происходит?) во время Мукополисахаридоза типа I-Н (синдрома Гурлера)
Патоморфологическая картина:
наблюдается

Нарушения при МПС I-H:
Нарушения при МПС I-H:


Особенности фенотипа.
Характерны изменения черт лица по типу «гаргоилизма», которые становятся
Особенности фенотипа.
Характерны изменения черт лица по типу «гаргоилизма», которые становятся
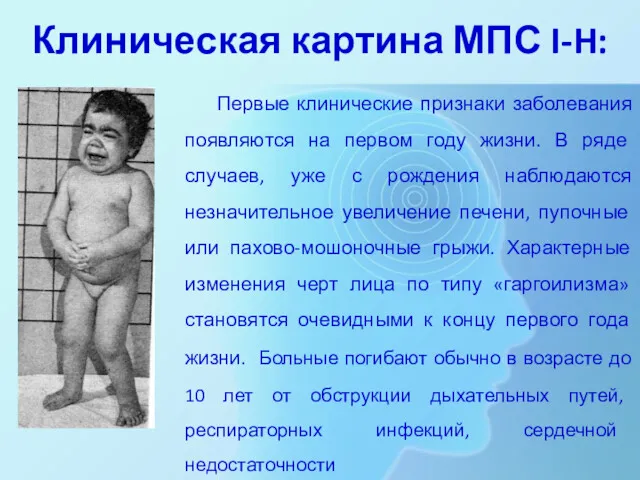
Клиническая картина МПС I-H:
Первые клинические признаки заболевания появляются на первом
Клиническая картина МПС I-H:
Первые клинические признаки заболевания появляются на первом

Клиническая диагностика
В ряде случаев, выраженный клинический полиморфизм МПС I приводит
Клиническая диагностика
В ряде случаев, выраженный клинический полиморфизм МПС I приводит

Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера):
Подтверждающая биохимическая диагностика МПС I заключается
Диагностика Мукополисахаридоза типа I-Н (синдрома Гурлера):
Подтверждающая биохимическая диагностика МПС I заключается

Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера):
Заменительная терапия.
Трансплантация стволовых клеток.
Хирургическая коррекция глаукомы,
Лечение Мукополисахаридоза типа I-Н (синдрома Гурлера):
Заменительная терапия.
Трансплантация стволовых клеток.
Хирургическая коррекция глаукомы,
 Cardiogenic shock
Cardiogenic shock Применение статистики для оценки здоровья. Лекция 1
Применение статистики для оценки здоровья. Лекция 1 Личностные расстройства и акцентуации характера
Личностные расстройства и акцентуации характера Биологические ритмы и работоспособность
Биологические ритмы и работоспособность Постгистерэктомический синдром
Постгистерэктомический синдром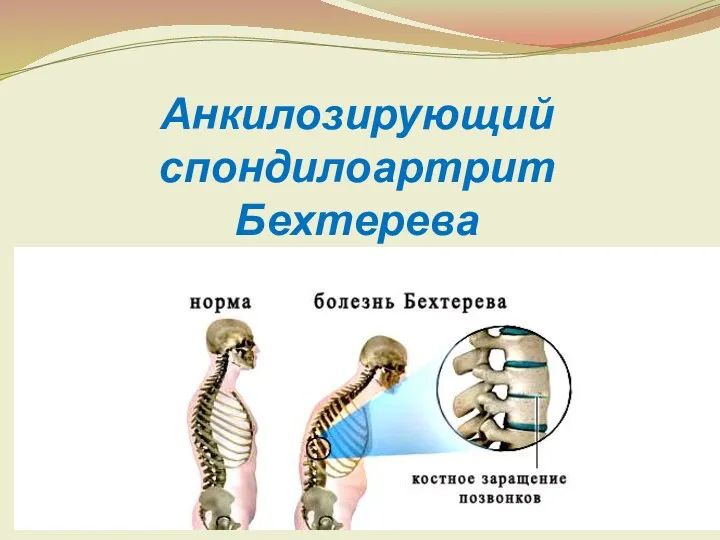 Анкилозирующий спондилоартрит Бехтерева
Анкилозирующий спондилоартрит Бехтерева Острые респираторные инфекции. Пневмония
Острые респираторные инфекции. Пневмония Послеродовое гипотоническое кровотечение: рекомендации ВОЗ по оказанию медицинской помощи
Послеродовое гипотоническое кровотечение: рекомендации ВОЗ по оказанию медицинской помощи Дистресс плода
Дистресс плода Инвалидность и ее причины, реабилитация
Инвалидность и ее причины, реабилитация Пневмонии и рак легких
Пневмонии и рак легких Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою
Туберкулезге қарсы препараттар, фармакокинетикасы, фармакодинамикасы, жанама әсерлері және оларды жою Анемиялық синдром. Этиологиясы. Классификациясы. Патоморфологиялық сипаттамасы
Анемиялық синдром. Этиологиясы. Классификациясы. Патоморфологиялық сипаттамасы Эпидемиологическая ситуация по ВИЧ/СПИДу в мире и в России
Эпидемиологическая ситуация по ВИЧ/СПИДу в мире и в России Локальні протоколи в медичній реабілітації
Локальні протоколи в медичній реабілітації Лечебное применение механических факторов (часть 2)
Лечебное применение механических факторов (часть 2) Дизартрия, коррекция. 1 этап
Дизартрия, коррекция. 1 этап Занятие №3. Особенности развития нервной системы детей
Занятие №3. Особенности развития нервной системы детей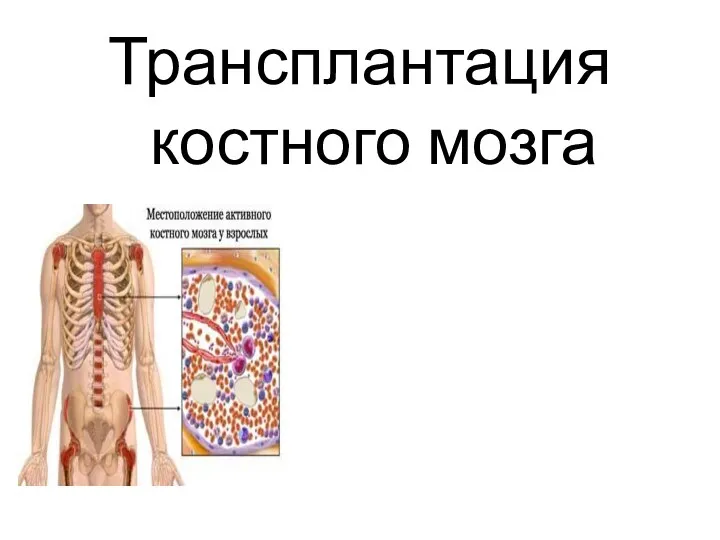 Трансплантация костного мозга
Трансплантация костного мозга Первичная медико-санитарная помощь
Первичная медико-санитарная помощь Ішкі жұқпалы емес аурулар клиникалық диагностикамен. Ветеринариялық зоогигиена, хирургия
Ішкі жұқпалы емес аурулар клиникалық диагностикамен. Ветеринариялық зоогигиена, хирургия Аномалії кісткового таза
Аномалії кісткового таза Применение лекарственных средств, используемых при сердечно-сосудистых заболеваниях у беременных и родильниц
Применение лекарственных средств, используемых при сердечно-сосудистых заболеваниях у беременных и родильниц Органы нервной системы. Спинной и головной мозг. Рефлекторные дуги. Гистология
Органы нервной системы. Спинной и головной мозг. Рефлекторные дуги. Гистология Жедел улану. Антидотты терапия
Жедел улану. Антидотты терапия Проблемы ранней беременности у подростков
Проблемы ранней беременности у подростков Гигиена. Структура окружающей среды. Концепция факторов риска. Модели развития главных неинфекционных болезней, их профилактика
Гигиена. Структура окружающей среды. Концепция факторов риска. Модели развития главных неинфекционных болезней, их профилактика Лекция №5. Тема 1.4. Организация и проведение работы в центрах (отделениях) медицинской профилактики, центрах здоровья
Лекция №5. Тема 1.4. Организация и проведение работы в центрах (отделениях) медицинской профилактики, центрах здоровья