- Наследственные болезни обмена веществ: клиника, диагностика, лечение

Содержание
- 2. Наследственные болезни обмена веществ Обширный класс моногенных заболеваний, обусловленных мутациями в генах, кодирующих: - ферменты (чаще)
- 3. Биохимические маркеры заболевания в десятки раз отличаются от нормы Известно около 500 нозологических форм Каждая из
- 4. Классификация 22 подкласса в зависимости от пораженного метаболического пути Подклассы: Частота Аминоацидопатии 31% Органические ацидурии 27%
- 5. Аутосомно-рецессивный тип наследования Фенилкетонурия 1:8 000 Болезнь Тея-Сакса 1:120 000 (среди евреев-ашкенази) 1:3 000 Болезнь Гоше
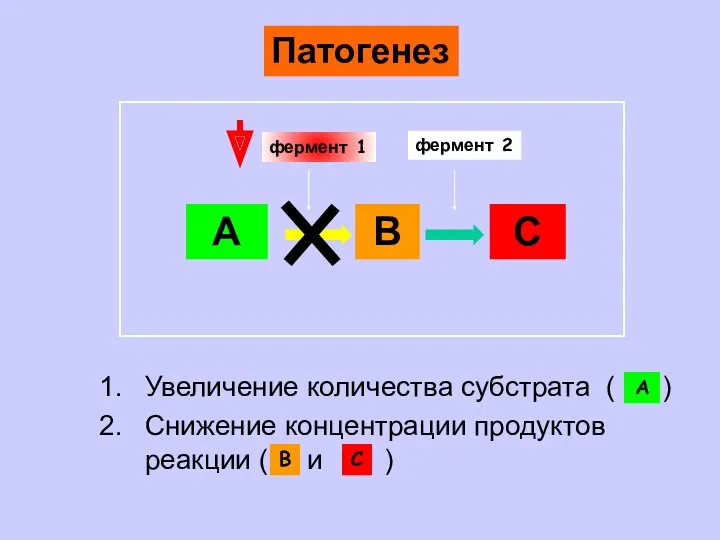
- 6. А В С фермент 1 фермент 2 Увеличение количества субстрата ( ) Снижение концентрации продуктов реакции
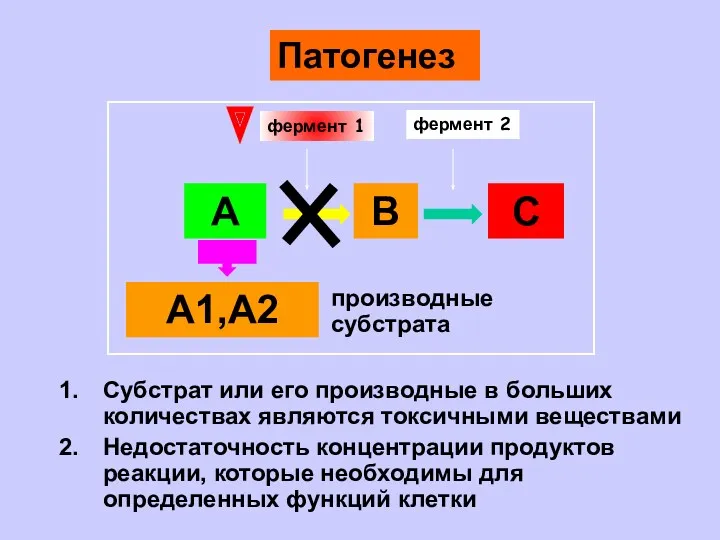
- 7. А В С А1,А2 фермент 1 фермент 2 Субстрат или его производные в больших количествах являются
- 8. Наследственные болезни обмена веществ Типы наследования Аутосомно-рецессивный (подавляющее большинство форм) 2. Х-сцепленный рецессивный 3. Митохондриальный (цитоплазматический)
- 9. Диагностика
- 10. Анамнез жизни и заболевания Дебют заболевания после различного по продолжительности относительно нормального развития Начало болезни связано
- 11. Неонатальный период (0-1 месяц) Дебют заболевания от 24 часов до нескольких недель Острое начало болезни и
- 12. Неонатальный период. неврологические расстройства Органические ацидурии (Метилмалоновая, пропионовая) Дефекты цикла мочевины Болезнь с запахом кленового сиропа
- 13. Неонатальный период. Ведущие симптомы –поражение печени Галактоземия Тирозинемия Недостаточность а1-антитрипсина Синдром Цельвейгера Болезнь Ниманна-Пика тип С
- 14. Детский возраст (6 месяцев - 5 лет) Дебют заболевания после различного по продолжительности относительно нормального развития
- 15. Детский возраст Лизосомные болезни накопления Митохондриальные болезни Некоторые органические ацидурии Нарушения углеводного обмена ( гликогенозы, фруктоземия)
- 16. Юношеский возраст Прогрессирующий характер течения болезни Задержка психического развития, снижение школьной успеваемости Аутистическое поведение и/или эпизоды
- 17. Юношеский возраст Митохондриальные болезни Лизосомные болезни Пероксисомные болезни
- 18. Лабораторные исследования Клинический анализ крови (тробоцитопения, лейкопения, анемия , ретикулоцитоз) Клинический анализ мочи (изменение цвета, необычный
- 19. Специфический запах мочи и тела «Сладкий» ( запах карамели, кленового сиропа) – Лейциноз «Вареной капусты» –
- 20. Фенотип Большинство больных с НБО имеют обычный фенотип Специфические особенности фенотипа могут служить важным ключом для
- 21. Особенности фенотипа
- 22. Лизосомные болезни накопления Мукополисахаридозы Муколипидозы α-маннозидоз GM1-ганглиозидоз Галактосиалидоз
- 23. Синдром Гурлер (МПС тип I)
- 24. from Metabolic Basis of Inherited Disease 7th ed
- 25. GM1-ганглиозидоз Собственные наблюдения
- 26. Мукополисахаридоз тип IV from Metabolic Basis of Inherited Disease 7th ed
- 27. Пероксисомные болезни Синдром Цельвейгера Инфантильная болезнь Рефсума Неонатальная адренолейкодистрофия Ризомелическая точечная хондродисплазия
- 28. Синдром Цельвейгера
- 29. Гепато- и/или спленомегалия Лизосомные болезни накопления Болезнь Гоше, болезнь Ниманна-Пика А/В/С, болезнь Вольмана, ганглиозидозы, а-маннозидоз, галактосиалидоз,
- 30. Изменения кожи и придатков кожи Алопеция Жесткие волосы Курчавые волосы Ангиокератома Дерматит Ихтиоз
- 31. Недостаточность биотинидазы Собственные наблюдения
- 32. Болезнь Менкенса
- 33. Ангиокератома Болезнь Фабри Фукозидоз Сиалидоз Ганглиозидозы
- 34. Ихтиоз Болезнь Съергена-Ларсона Болезнь Рефсума (поздняя форма) Множественная сульфатазная недостаточность
- 35. Нарушения органа зрения Катаракта Глаукома Подвывих/вывих хрусталика Пигментная дегенерация сетчатки Атрофия зрительных нервов Дегенерация макулы Кольцо
- 36. Дегенерация макулы по типу «вишневой косточки»
- 37. Дегенерация макулы по типу «вишневой косточки» Заболевания Встречаемость Сиалидоз Почти всегда GM-2 ганглиозидозы Почти всегда Нимана-Пика
- 38. Кольцо Кайзера-Флейшера
- 39. Катаракта Лизосомные болезни накопления Альфа маннозидоз, галактосиалидоз, сиалидоз Пероксисомные заболевания Синдром Цельвейгера, болезнь Рефсума Митохондриальные болезни
- 40. МРТ/КТ головного мозга Высокоспецифичны для лейкодистрофий, болезни Лея, болезни Вильсона-Коновалова, болезни Галлервордена-Шпатца Для большинства НБО –
- 41. Лейкодистрофии
- 42. Метахроматическая лейкодистрофия Собственные наблюдения
- 43. Болезнь Канавана Собственные наблюдения
- 44. Х-сцепленная адренолейкодистрофия Собственные наблюдения
- 45. Поражение подкорковых структур
- 46. Болезнь Вильсона-Коновалова
- 47. Синдром Лея Собственные наблюдения
- 48. Болезнь Галлервордена-Шпатца
- 49. Другие изменения
- 50. Глутаровая ацидурия тип 1 American J.Med Genetics121C:38–52 (2003)
- 51. Основные методы точной диагностики НБО Биохимические Молекулярно- генетические
- 52. Методические подходы к диагностике Ген Белок Метаболиты ДНК-диагностика Энзимодиагностика и другие методы анализа белков Хроматографические или
- 53. Диагностика на уровне метаболитов Плазма крови Цереброспинальная жидкость Моча Пятна высушенной крови Иногда необходимо проведение нагрузочных
- 54. Диагностика на уровне метаболитов Органические ацидурии Аминоацидопатии Мукополисахаридозы Дефекты митохондриального в-окисления Пероксисомные болезни
- 55. Определение активности ферментов Биологический материал: лейкоциты периферической крови, плазма крови, культура кожных фибробластов, пренатальная диагностика –
- 56. Определение активности ферментов Лизосомные болезни накопления Митохондриальные болени Гликогенозы Некоторые органические ацидурии и аминоацидопатии
- 57. ДНК-диагностика Диагностика носительства заболеваний Диагностика заболеваний с неизвестным первичным биохимическим дефектом Диагностика заболеваний при которых биохимические
- 58. А В С А1,А2 фермент 1 фермент 2 Выведение токсичных метаболитов Ограничение поступления субстрата Восполнение недостающего
- 59. Москва, ул. Москворечье д.1, 101,103 Тел. (495) 324 2004 labnbo@med-gen.ru labnbo@yandex.ru Лаборатория наследственных болезней обмена веществ
- 61. Скачать презентацию

Наследственные болезни обмена веществ
Обширный класс моногенных заболеваний, обусловленных мутациями в генах,
Наследственные болезни обмена веществ
Обширный класс моногенных заболеваний, обусловленных мутациями в генах,

Биохимические маркеры заболевания в десятки раз отличаются от нормы
Известно около
Биохимические маркеры заболевания в десятки раз отличаются от нормы
Известно около

Классификация
22 подкласса в зависимости от пораженного метаболического пути
Подклассы: Частота
Аминоацидопатии 31%
Органические ацидурии
Классификация
22 подкласса в зависимости от пораженного метаболического пути Подклассы: Частота Аминоацидопатии 31% Органические ацидурии

Аутосомно-рецессивный тип наследования
Фенилкетонурия 1:8 000
Болезнь Тея-Сакса 1:120 000
(среди евреев-ашкенази) 1:3 000
Болезнь Гоше 1:40 000
Болезнь
Аутосомно-рецессивный тип наследования Фенилкетонурия 1:8 000 Болезнь Тея-Сакса 1:120 000 (среди евреев-ашкенази) 1:3 000 Болезнь Гоше 1:40 000 Болезнь
А
В
С
фермент 1
фермент 2
Увеличение количества субстрата ( )
Снижение концентрации продуктов
А
В
С
фермент 1
фермент 2
Увеличение количества субстрата ( )
Снижение концентрации продуктов
А
В
С
А1,А2
фермент 1
фермент 2
Субстрат или его производные в больших количествах являются
А
В
С
А1,А2
фермент 1
фермент 2
Субстрат или его производные в больших количествах являются

Наследственные болезни обмена веществ
Типы наследования
Аутосомно-рецессивный
(подавляющее большинство форм)
2. Х-сцепленный рецессивный
3.
Наследственные болезни обмена веществ
Типы наследования
Аутосомно-рецессивный
(подавляющее большинство форм)
2. Х-сцепленный рецессивный
3.

Диагностика
Диагностика

Анамнез жизни и заболевания
Дебют заболевания после различного по продолжительности относительно нормального
Анамнез жизни и заболевания
Дебют заболевания после различного по продолжительности относительно нормального

Неонатальный период
(0-1 месяц)
Дебют заболевания от 24 часов до нескольких
Неонатальный период
(0-1 месяц)
Дебют заболевания от 24 часов до нескольких

Неонатальный период. неврологические расстройства
Органические ацидурии (Метилмалоновая, пропионовая)
Дефекты цикла мочевины
Болезнь с запахом
Неонатальный период. неврологические расстройства
Органические ацидурии (Метилмалоновая, пропионовая)
Дефекты цикла мочевины
Болезнь с запахом

Неонатальный период. Ведущие симптомы –поражение печени
Галактоземия
Тирозинемия
Недостаточность а1-антитрипсина
Синдром Цельвейгера
Болезнь Ниманна-Пика тип С
Гликогеноз
Неонатальный период. Ведущие симптомы –поражение печени
Галактоземия
Тирозинемия
Недостаточность а1-антитрипсина
Синдром Цельвейгера
Болезнь Ниманна-Пика тип С
Гликогеноз

Детский возраст
(6 месяцев - 5 лет)
Дебют заболевания после различного по
Детский возраст
(6 месяцев - 5 лет)
Дебют заболевания после различного по

Детский возраст
Лизосомные болезни накопления
Митохондриальные болезни
Некоторые органические ацидурии
Нарушения углеводного обмена ( гликогенозы,
Детский возраст
Лизосомные болезни накопления
Митохондриальные болезни
Некоторые органические ацидурии
Нарушения углеводного обмена ( гликогенозы,

Юношеский возраст
Прогрессирующий характер течения болезни
Задержка психического развития, снижение школьной успеваемости
Аутистическое поведение
Юношеский возраст
Прогрессирующий характер течения болезни
Задержка психического развития, снижение школьной успеваемости
Аутистическое поведение

Юношеский возраст
Митохондриальные болезни
Лизосомные болезни
Пероксисомные болезни
Юношеский возраст
Митохондриальные болезни
Лизосомные болезни
Пероксисомные болезни

Лабораторные исследования
Клинический анализ крови (тробоцитопения, лейкопения, анемия , ретикулоцитоз)
Клинический анализ мочи
Лабораторные исследования
Клинический анализ крови (тробоцитопения, лейкопения, анемия , ретикулоцитоз)
Клинический анализ мочи

Специфический запах мочи и тела
«Сладкий» ( запах карамели, кленового сиропа) –
Специфический запах мочи и тела
«Сладкий» ( запах карамели, кленового сиропа) –

Фенотип
Большинство больных с НБО имеют обычный фенотип
Специфические особенности фенотипа могут
Фенотип
Большинство больных с НБО имеют обычный фенотип
Специфические особенности фенотипа могут

Особенности фенотипа
Особенности фенотипа

Лизосомные болезни накопления
Мукополисахаридозы
Муколипидозы
α-маннозидоз
GM1-ганглиозидоз
Галактосиалидоз
Лизосомные болезни накопления
Мукополисахаридозы
Муколипидозы
α-маннозидоз
GM1-ганглиозидоз
Галактосиалидоз

Синдром Гурлер (МПС тип I)
Синдром Гурлер (МПС тип I)
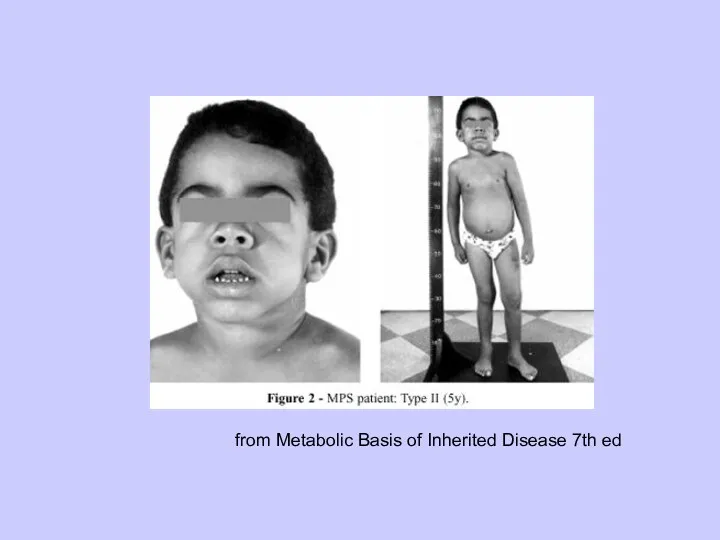
from Metabolic Basis of Inherited Disease 7th ed
from Metabolic Basis of Inherited Disease 7th ed
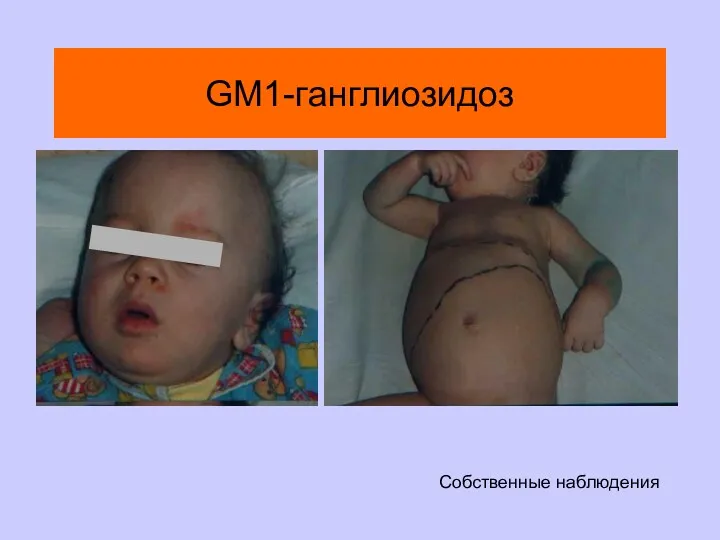
GM1-ганглиозидоз
Собственные наблюдения
GM1-ганглиозидоз
Собственные наблюдения
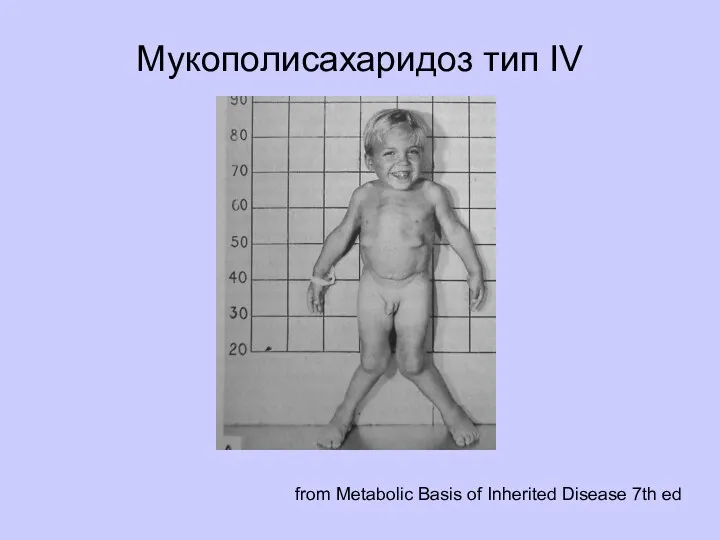
Мукополисахаридоз тип IV
from Metabolic Basis of Inherited Disease 7th ed
Мукополисахаридоз тип IV
from Metabolic Basis of Inherited Disease 7th ed

Пероксисомные болезни
Синдром Цельвейгера
Инфантильная болезнь Рефсума
Неонатальная адренолейкодистрофия
Ризомелическая точечная хондродисплазия
Пероксисомные болезни
Синдром Цельвейгера
Инфантильная болезнь Рефсума
Неонатальная адренолейкодистрофия
Ризомелическая точечная хондродисплазия
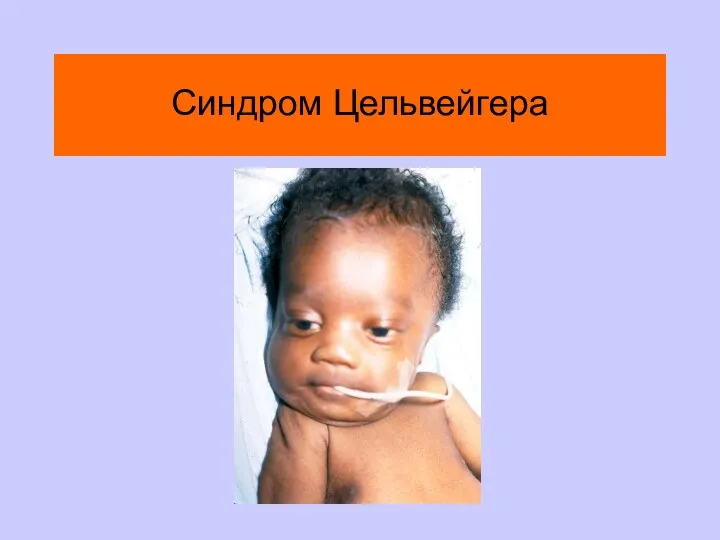
Синдром Цельвейгера
Синдром Цельвейгера

Гепато- и/или спленомегалия
Лизосомные болезни накопления
Болезнь Гоше, болезнь Ниманна-Пика А/В/С, болезнь Вольмана,
Гепато- и/или спленомегалия
Лизосомные болезни накопления
Болезнь Гоше, болезнь Ниманна-Пика А/В/С, болезнь Вольмана,

Изменения кожи и придатков кожи
Алопеция
Жесткие волосы
Курчавые волосы
Ангиокератома
Дерматит
Ихтиоз
Изменения кожи и придатков кожи
Алопеция
Жесткие волосы
Курчавые волосы
Ангиокератома
Дерматит
Ихтиоз

Недостаточность биотинидазы
Собственные наблюдения
Недостаточность биотинидазы
Собственные наблюдения

Болезнь Менкенса
Болезнь Менкенса
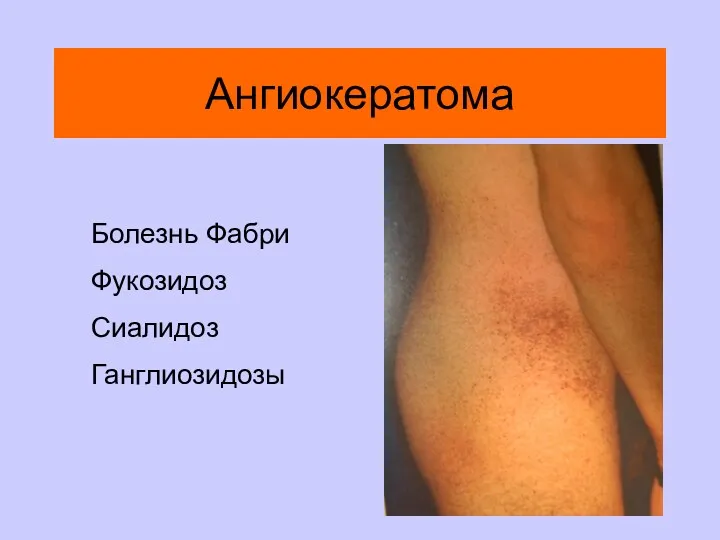
Ангиокератома
Болезнь Фабри
Фукозидоз
Сиалидоз
Ганглиозидозы
Ангиокератома
Болезнь Фабри
Фукозидоз
Сиалидоз
Ганглиозидозы

Ихтиоз
Болезнь Съергена-Ларсона
Болезнь Рефсума (поздняя форма)
Множественная сульфатазная
недостаточность
Ихтиоз
Болезнь Съергена-Ларсона
Болезнь Рефсума (поздняя форма)
Множественная сульфатазная
недостаточность

Нарушения органа зрения
Катаракта
Глаукома
Подвывих/вывих хрусталика
Пигментная дегенерация сетчатки
Атрофия зрительных нервов
Дегенерация макулы
Кольцо Кайзера-Флейшнера
Нарушения органа зрения
Катаракта
Глаукома
Подвывих/вывих хрусталика
Пигментная дегенерация сетчатки
Атрофия зрительных нервов
Дегенерация макулы
Кольцо Кайзера-Флейшнера
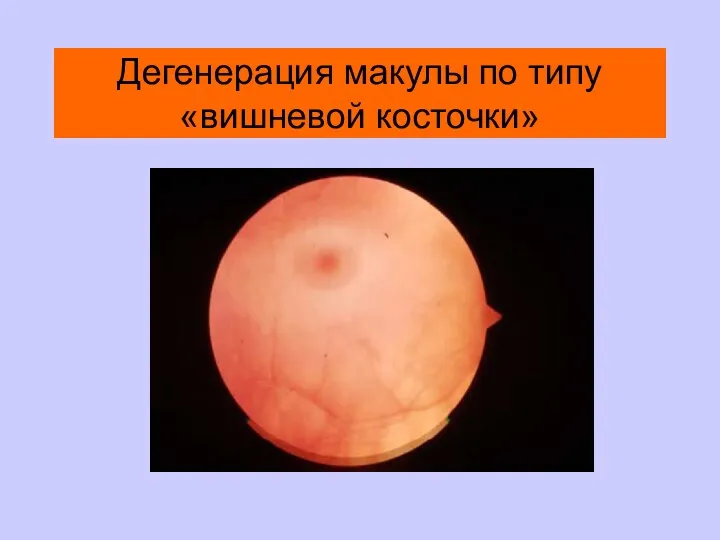
Дегенерация макулы по типу «вишневой косточки»
Дегенерация макулы по типу «вишневой косточки»

Дегенерация макулы по типу «вишневой косточки»
Заболевания Встречаемость
Сиалидоз Почти всегда
GM-2 ганглиозидозы Почти всегда
Нимана-Пика тип
Дегенерация макулы по типу «вишневой косточки»
Заболевания Встречаемость
Сиалидоз Почти всегда
GM-2 ганглиозидозы Почти всегда
Нимана-Пика тип
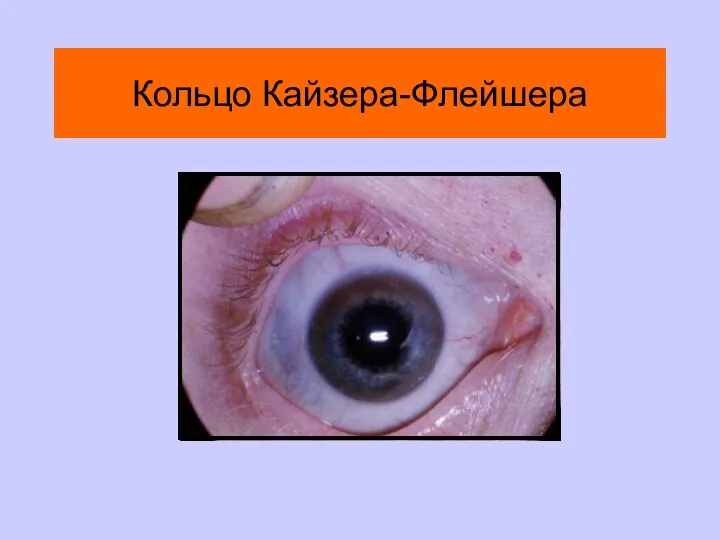
Кольцо Кайзера-Флейшера
Кольцо Кайзера-Флейшера

Катаракта
Лизосомные болезни накопления
Альфа маннозидоз, галактосиалидоз, сиалидоз
Пероксисомные заболевания
Синдром Цельвейгера, болезнь Рефсума
Митохондриальные болезни
Нарушения
Катаракта
Лизосомные болезни накопления
Альфа маннозидоз, галактосиалидоз, сиалидоз
Пероксисомные заболевания
Синдром Цельвейгера, болезнь Рефсума
Митохондриальные болезни
Нарушения

МРТ/КТ головного мозга
Высокоспецифичны для лейкодистрофий, болезни Лея, болезни Вильсона-Коновалова, болезни Галлервордена-Шпатца
Для
МРТ/КТ головного мозга
Высокоспецифичны для лейкодистрофий, болезни Лея, болезни Вильсона-Коновалова, болезни Галлервордена-Шпатца
Для

Лейкодистрофии
Лейкодистрофии
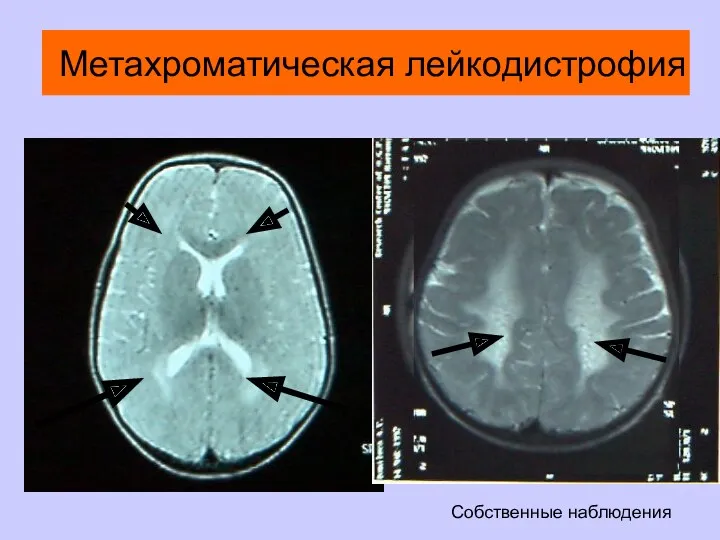
Метахроматическая лейкодистрофия
Собственные наблюдения
Метахроматическая лейкодистрофия
Собственные наблюдения
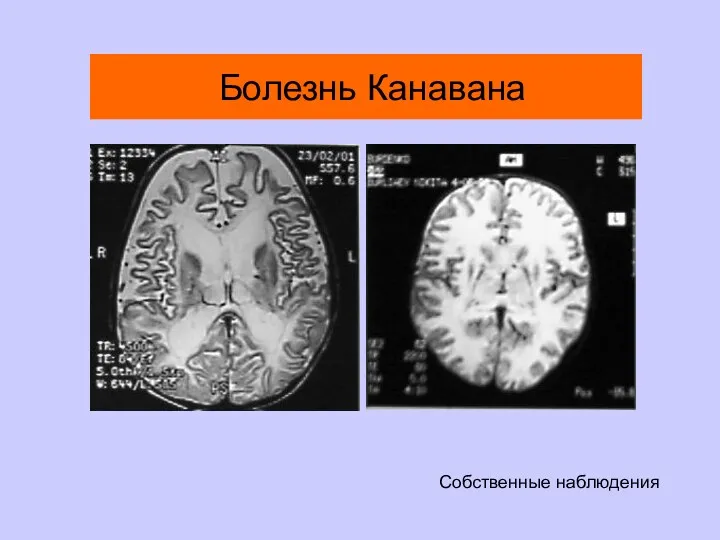
Болезнь Канавана
Собственные наблюдения
Болезнь Канавана
Собственные наблюдения
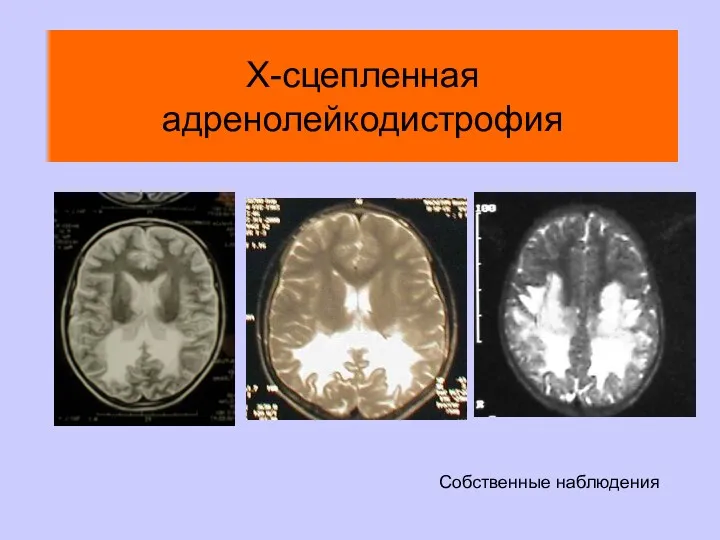
Х-сцепленная адренолейкодистрофия
Собственные наблюдения
Х-сцепленная адренолейкодистрофия
Собственные наблюдения

Поражение подкорковых структур
Поражение подкорковых структур
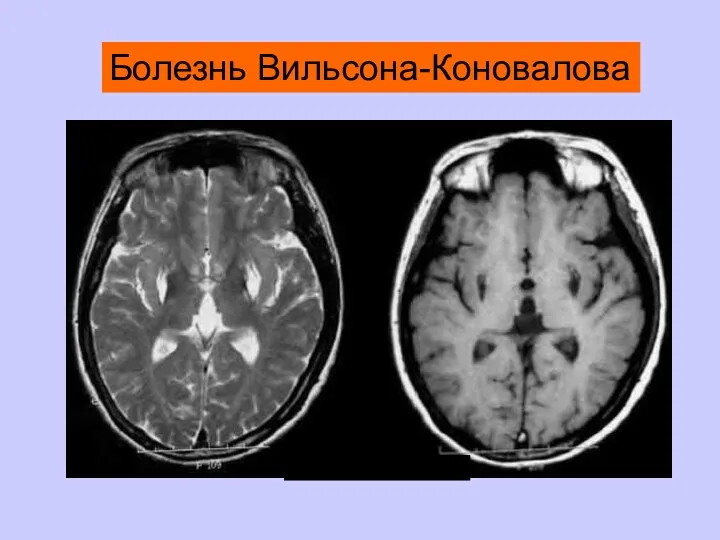
Болезнь Вильсона-Коновалова
Болезнь Вильсона-Коновалова
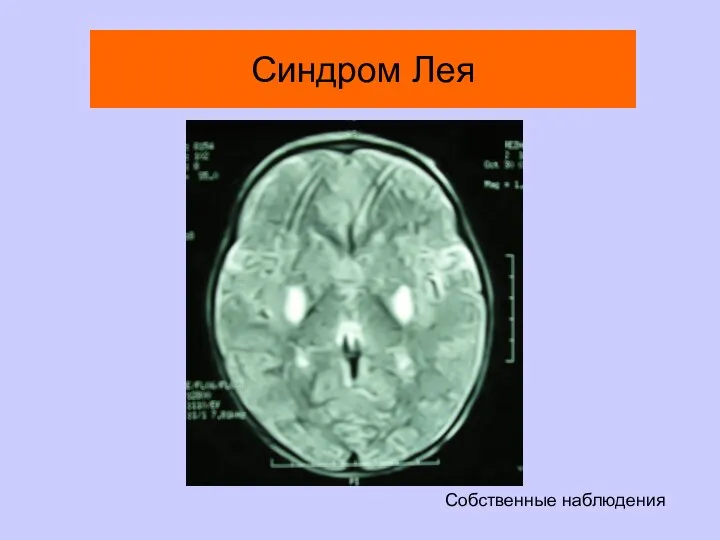
Синдром Лея
Собственные наблюдения
Синдром Лея
Собственные наблюдения
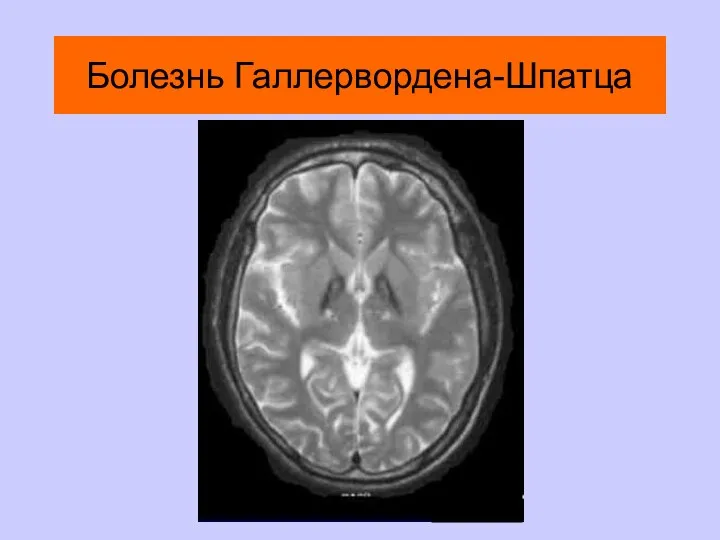
Болезнь Галлервордена-Шпатца
Болезнь Галлервордена-Шпатца

Другие изменения
Другие изменения
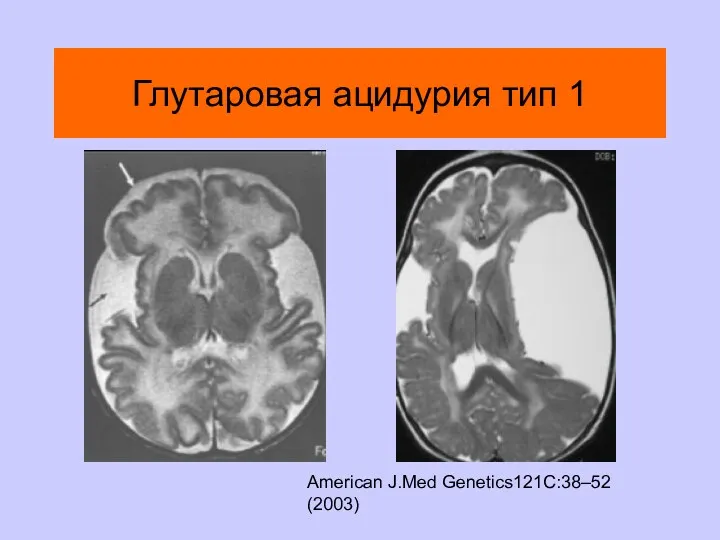
Глутаровая ацидурия тип 1
American J.Med Genetics121C:38–52 (2003)
Глутаровая ацидурия тип 1
American J.Med Genetics121C:38–52 (2003)

Основные методы точной диагностики НБО
Биохимические
Молекулярно- генетические
Основные методы точной диагностики НБО
Биохимические
Молекулярно- генетические
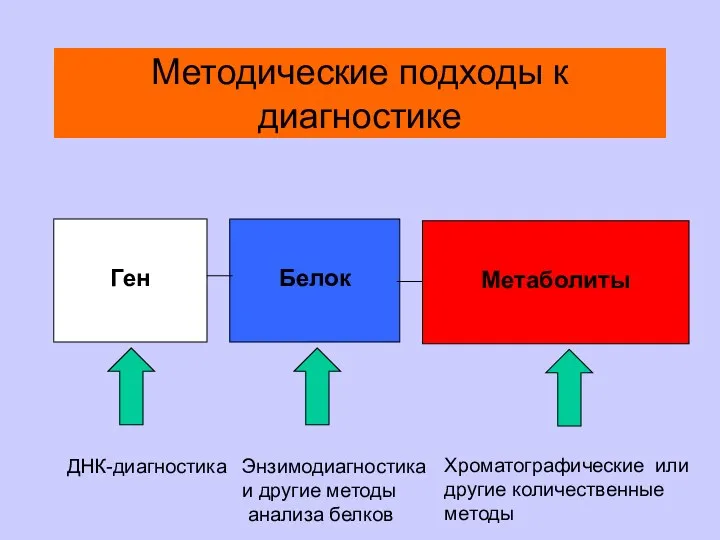
Методические подходы к диагностике
Ген
Белок
Метаболиты
ДНК-диагностика
Энзимодиагностика
и другие методы
анализа белков
Хроматографические или
другие
Методические подходы к диагностике
Ген
Белок
Метаболиты
ДНК-диагностика
Энзимодиагностика
и другие методы
анализа белков
Хроматографические или
другие

Диагностика на уровне метаболитов
Плазма крови
Цереброспинальная жидкость
Моча
Пятна высушенной крови
Иногда необходимо проведение нагрузочных
Диагностика на уровне метаболитов
Плазма крови
Цереброспинальная жидкость
Моча
Пятна высушенной крови
Иногда необходимо проведение нагрузочных

Диагностика на уровне метаболитов
Органические ацидурии
Аминоацидопатии
Мукополисахаридозы
Дефекты митохондриального в-окисления
Пероксисомные болезни
Диагностика на уровне метаболитов
Органические ацидурии
Аминоацидопатии
Мукополисахаридозы
Дефекты митохондриального в-окисления
Пероксисомные болезни

Определение активности ферментов
Биологический материал: лейкоциты периферической крови, плазма крови, культура кожных
Определение активности ферментов
Биологический материал: лейкоциты периферической крови, плазма крови, культура кожных

Определение активности ферментов
Лизосомные болезни накопления
Митохондриальные болени
Гликогенозы
Некоторые органические ацидурии и аминоацидопатии
Определение активности ферментов
Лизосомные болезни накопления
Митохондриальные болени
Гликогенозы
Некоторые органические ацидурии и аминоацидопатии

ДНК-диагностика
Диагностика носительства заболеваний
Диагностика заболеваний с неизвестным первичным биохимическим дефектом
Диагностика заболеваний
ДНК-диагностика
Диагностика носительства заболеваний
Диагностика заболеваний с неизвестным первичным биохимическим дефектом
Диагностика заболеваний
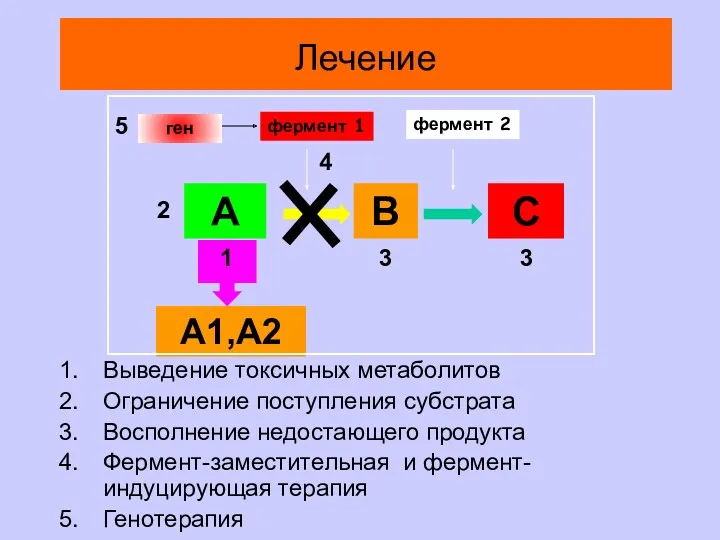
А
В
С
А1,А2
фермент 1
фермент 2
Выведение токсичных метаболитов
Ограничение поступления субстрата
Восполнение недостающего продукта
Фермент-заместительная и
А
В
С
А1,А2
фермент 1
фермент 2
Выведение токсичных метаболитов
Ограничение поступления субстрата
Восполнение недостающего продукта
Фермент-заместительная и

Москва, ул. Москворечье д.1, 101,103
Тел. (495) 324 2004
labnbo@med-gen.ru
labnbo@yandex.ru
Лаборатория наследственных болезней обмена
Москва, ул. Москворечье д.1, 101,103
Тел. (495) 324 2004
labnbo@med-gen.ru
labnbo@yandex.ru
Лаборатория наследственных болезней обмена
 Заболевания щитовидной железы
Заболевания щитовидной железы Инновационные методы лечения головной боли
Инновационные методы лечения головной боли Вегетарианство. Вред или польза
Вегетарианство. Вред или польза Интоксикация фосфороорганическими пестицидами. Отравление окислами азота, сероводородом и сернистым ангидридом
Интоксикация фосфороорганическими пестицидами. Отравление окислами азота, сероводородом и сернистым ангидридом Дамудың қатерлі кезеңдері
Дамудың қатерлі кезеңдері Анти-акне MESOLAB®
Анти-акне MESOLAB® Осложнения лекарственной терапии. Побочные действия и токсические свойства лекарств
Осложнения лекарственной терапии. Побочные действия и токсические свойства лекарств Доказательная медицина & Клинический подход
Доказательная медицина & Клинический подход Структура ФБУЗ Центр гигиены и эпидемиологии в городе Москве
Структура ФБУЗ Центр гигиены и эпидемиологии в городе Москве Конструкции различных видов протезов на дентальных имплантатах
Конструкции различных видов протезов на дентальных имплантатах Нейроэндокринные синдромы у женщин
Нейроэндокринные синдромы у женщин Координаторная система. Мозжечок, синдромы поражения. Экстрапирамидная система, синдромы поражения
Координаторная система. Мозжечок, синдромы поражения. Экстрапирамидная система, синдромы поражения Фекально-оральный механизм передачи инфекции
Фекально-оральный механизм передачи инфекции Организационно-правовые аспекты оказания первой помощи
Организационно-правовые аспекты оказания первой помощи АФО органов пищеварения у детей
АФО органов пищеварения у детей Токсоплазмоз. Роль домашних животных
Токсоплазмоз. Роль домашних животных Бауырдың жедел зақымдалуының патофизиологиясы. Жедел гепатиттер сиппатамасы. Бауырдың созылмалы зақымдалуының патофизиологиясы
Бауырдың жедел зақымдалуының патофизиологиясы. Жедел гепатиттер сиппатамасы. Бауырдың созылмалы зақымдалуының патофизиологиясы Клинико-психолого-педагогическая характеристика детей с нарушениями речи
Клинико-психолого-педагогическая характеристика детей с нарушениями речи Аномалии развития и положения женских половых органов
Аномалии развития и положения женских половых органов Форменные элементы крови
Форменные элементы крови Балалардың асқазан - ішек аурулары туралы түсінік беру. Балаларда ішек жұқпасының көріністері
Балалардың асқазан - ішек аурулары туралы түсінік беру. Балаларда ішек жұқпасының көріністері Заманауи гепатопротекторлы заттар
Заманауи гепатопротекторлы заттар Туляремия
Туляремия Маркировка. Лекарства. Качество и Безопасность
Маркировка. Лекарства. Качество и Безопасность Рак молочной железы (РМЖ)
Рак молочной железы (РМЖ) Геморрой. Предрасполагающие факторы
Геморрой. Предрасполагающие факторы Исследование отделяемого половых органов
Исследование отделяемого половых органов Әскери медицина туралы түсінік. Медицина қызметін ұйымдастыру және тактикасы, ғылыми тұрғыда және пән ретінде оқып үйрену
Әскери медицина туралы түсінік. Медицина қызметін ұйымдастыру және тактикасы, ғылыми тұрғыда және пән ретінде оқып үйрену