- Шляхи і проблеми в створенні оригінальних лікарських засобів в Україні

Содержание
- 2. Оригінальні лікарські засоби У зв’язку з динамічним розвитком фармацевтичного ринку дедалі частіше постає питання щодо оригінальних
- 3. Розробка та впровадження Процес розробки та впровадження нового лікарського препарату потребує участі багатьох висококваліфікованих спеціалістів, великих
- 4. Суб’єкти створення лікарських засобів Лікарські засоби можуть створюватись підприємствами, установами, організаціями та громадянами. Автором лікарського засобу
- 5. Етапи створення нових лікарських засобів Першим етапом є пошук та отримання нових речовин (синтезованих або виділених
- 6. Доклінічні дослідження Наступний етап доклінічного вивчення передбачає: • з’ясування специфічної фармакологічної дії на різних моделях захворювань
- 7. Клінічні випробування Якщо результати доклінічних досліджень, на етапі яких майбутні ліки мають назву «фармакологічних препаратів», переконують
- 8. Препарат має затверджену при реєстрації міжнародну непатентовану назву (МНН) і торгову (фірмову) назву, під якою його
- 9. Генериком (дженериком) називається непатентований лікарський препарат, який є відтворенням оригінального препарату, на діючі речовини якого збіг
- 10. Центральним питанням проблеми порівняння генериків із оригінальним препаратом є гарантована взаємозамінність. Вона, у свою чергу, базується
- 11. Фармакокінетична еквівалентність (біоеквівалентність), або ступінь схожості за фармакокінетичними параметрами фармацевтично еквівалентного препарату з референтним, означає, що
- 12. Роздрібні ціни на препарати амлодипіну станом на 01.11.2016 р
- 13. Актуальність проблеми вартості лікарських препаратів для України не потребує обговорення. Так, згідно з результатами соціологічного опитування,
- 14. ЗАКОН УКРАЇНИ «Про лікарські засоби” № 124/96-ВР від 04.04.96, ВВР, 1996, № 22, ст. 87 Розділ
- 15. В рамках НВО «Фарматрон» вперше в Україні налагоджено промисловий випуск «Тіотріазоліну» в таблетках, ампульних розчинах, ін’єкційних
- 16. Державні та міжнародні стандарти якості, як основа запобігання розповсюдженню неякісних та фальсифікованих лікарських засобів У відповідності
- 17. Стандартизація лікарських засобів - діяльність щодо встановлення правил, норм і характеристик для загального і багаторазового використання
- 18. Державна фармакопея України головний стандарт фармацевтичної галузі Державна Фармакопея України - Відповідно до Закону України «Про
- 19. Державна Фармакопея України 2.0 ДФУ 2 - це перша національна фармакопея України як повноправного члена ЄФ.
- 20. ДФУ 2.0 та Європейська фармакопея
- 21. Загальний обсяг ДФУ 2 перевищує 2000 сторінок, тому вона видається в трьох томах
- 23. Скачать презентацию

Оригінальні лікарські засоби
У зв’язку з динамічним розвитком фармацевтичного ринку дедалі частіше
Оригінальні лікарські засоби
У зв’язку з динамічним розвитком фармацевтичного ринку дедалі частіше

Розробка та впровадження
Процес розробки та впровадження нового лікарського препарату потребує участі
Розробка та впровадження
Процес розробки та впровадження нового лікарського препарату потребує участі

Суб’єкти створення лікарських засобів
Лікарські засоби можуть створюватись підприємствами, установами, організаціями та
Суб’єкти створення лікарських засобів Лікарські засоби можуть створюватись підприємствами, установами, організаціями та

Етапи створення нових лікарських засобів
Першим етапом є пошук та отримання
Етапи створення нових лікарських засобів
Першим етапом є пошук та отримання

Доклінічні дослідження
Наступний етап доклінічного вивчення передбачає:
• з’ясування специфічної фармакологічної дії на
Доклінічні дослідження
Наступний етап доклінічного вивчення передбачає: • з’ясування специфічної фармакологічної дії на

Клінічні випробування
Якщо результати доклінічних досліджень, на етапі яких майбутні ліки мають
Клінічні випробування
Якщо результати доклінічних досліджень, на етапі яких майбутні ліки мають

Препарат має затверджену при реєстрації міжнародну непатентовану назву (МНН) і торгову
Препарат має затверджену при реєстрації міжнародну непатентовану назву (МНН) і торгову

Генериком (дженериком) називається непатентований лікарський препарат, який є відтворенням оригінального препарату,
Генериком (дженериком) називається непатентований лікарський препарат, який є відтворенням оригінального препарату,

Центральним питанням проблеми порівняння генериків із оригінальним препаратом є гарантована взаємозамінність.
Центральним питанням проблеми порівняння генериків із оригінальним препаратом є гарантована взаємозамінність.

Фармакокінетична еквівалентність (біоеквівалентність), або ступінь схожості за фармакокінетичними параметрами фармацевтично еквівалентного
Фармакокінетична еквівалентність (біоеквівалентність), або ступінь схожості за фармакокінетичними параметрами фармацевтично еквівалентного

Роздрібні ціни на препарати амлодипіну станом на 01.11.2016 р
Роздрібні ціни на препарати амлодипіну станом на 01.11.2016 р

Актуальність проблеми вартості лікарських препаратів для України не потребує обговорення. Так,
Актуальність проблеми вартості лікарських препаратів для України не потребує обговорення. Так,

ЗАКОН УКРАЇНИ
«Про лікарські засоби”
№ 124/96-ВР від 04.04.96, ВВР, 1996, № 22,
ЗАКОН УКРАЇНИ «Про лікарські засоби” № 124/96-ВР від 04.04.96, ВВР, 1996, № 22,

В рамках НВО «Фарматрон» вперше в Україні налагоджено промисловий випуск «Тіотріазоліну»
В рамках НВО «Фарматрон» вперше в Україні налагоджено промисловий випуск «Тіотріазоліну»

Державні та міжнародні стандарти якості, як основа запобігання розповсюдженню неякісних та
Державні та міжнародні стандарти якості, як основа запобігання розповсюдженню неякісних та

Стандартизація лікарських засобів - діяльність щодо встановлення правил, норм і характеристик
Стандартизація лікарських засобів - діяльність щодо встановлення правил, норм і характеристик

Державна фармакопея України головний стандарт фармацевтичної галузі
Державна Фармакопея України - Відповідно
Державна фармакопея України головний стандарт фармацевтичної галузі Державна Фармакопея України - Відповідно

Державна Фармакопея України 2.0
ДФУ 2 - це перша національна фармакопея
Державна Фармакопея України 2.0
ДФУ 2 - це перша національна фармакопея
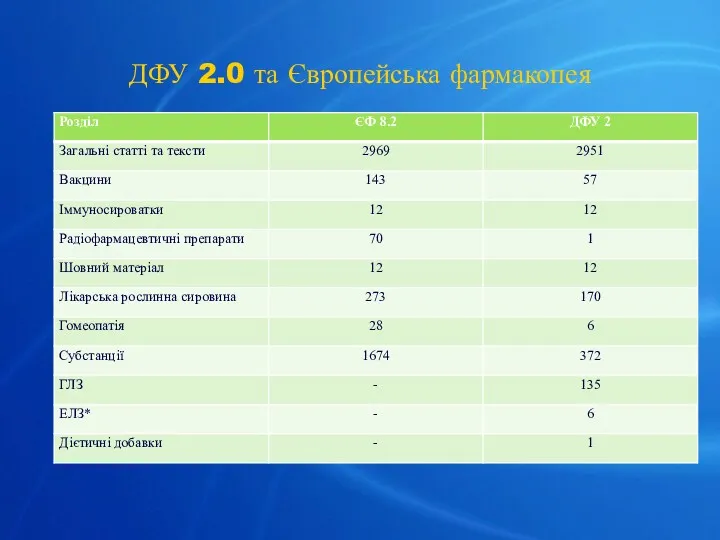
ДФУ 2.0 та Європейська фармакопея
ДФУ 2.0 та Європейська фармакопея

Загальний обсяг ДФУ 2 перевищує 2000 сторінок, тому вона видається в
Загальний обсяг ДФУ 2 перевищує 2000 сторінок, тому вона видається в
 Пропедевтика детских болезней
Пропедевтика детских болезней Симптомы, признаки и профилактика СПИДА
Симптомы, признаки и профилактика СПИДА Физиология выделения
Физиология выделения Балалардағы соматикалық аурулардың ауыз қуысындағы көріністері
Балалардағы соматикалық аурулардың ауыз қуысындағы көріністері Ортопедическое лечение
Ортопедическое лечение Несовместимые сочетания лекарственных веществ. Способы преодоления несовместимости
Несовместимые сочетания лекарственных веществ. Способы преодоления несовместимости Особенности вскармливания детей раннего возраста
Особенности вскармливания детей раннего возраста Лучевая диагностика органов ЖКТ
Лучевая диагностика органов ЖКТ Мастоидит, түрлері, клиникасы, диагностикасы, емі
Мастоидит, түрлері, клиникасы, диагностикасы, емі Гнойные заболевания легких и плевры
Гнойные заболевания легких и плевры Синдром слабости соединительной ткани
Синдром слабости соединительной ткани Фенотипические особенности наследственных синдромов рака молочной железы и яичников среди татарского этноса
Фенотипические особенности наследственных синдромов рака молочной железы и яичников среди татарского этноса Описание клинического случая
Описание клинического случая Основы реаниматологии
Основы реаниматологии Топографическая анатомия. Инструменты
Топографическая анатомия. Инструменты Заболеваемость населения Индивидуальность Физическое развитие. Лекция 2
Заболеваемость населения Индивидуальность Физическое развитие. Лекция 2 Первая доврачебная помощь при повреждениях и травмах
Первая доврачебная помощь при повреждениях и травмах Общая и функциональная артросиндесмология
Общая и функциональная артросиндесмология Предраковые заболевания. (Лекция 5)
Предраковые заболевания. (Лекция 5) Тоннельді егеп тазалау
Тоннельді егеп тазалау Лекция 8. Электрохимия
Лекция 8. Электрохимия Анатомия и биомеханика голеностопного сустава
Анатомия и биомеханика голеностопного сустава Неврология. Функции нервной системы. Спинной и головной мозг. Строение и функции
Неврология. Функции нервной системы. Спинной и головной мозг. Строение и функции Патология периода новорожденности
Патология периода новорожденности Фізіологічні основи оздоровчої фізичної культури
Фізіологічні основи оздоровчої фізичної культури Классификация, свойства и биологическая роль углеводов. (Тема 1)
Классификация, свойства и биологическая роль углеводов. (Тема 1) САП (malleus)
САП (malleus) Интерференцтерапия
Интерференцтерапия