- Спино-церебеллярная атаксия 1 типа

Содержание
- 2. Наследственные атаксии (НА) — клинически и генетически крайне гетерогенная группа наследственных нейродегенеративных заболеваний, основной характеристикой которых
- 3. Аутосомно-доминантные спиноцеребеллярные атаксии (АД-СЦА) характеризуются прогрессирующей атаксией, возникающей вследствие дегенерации мозжечка, а также, как правило, вовлечением
- 4. Первая классификация АД-СЦА, основанная на фенотипических характеристиках, была предложена A. Harding: она выделяла АД-СЦА типа I,
- 5. Хронологически первой формой АД-СЦА стала СЦА 1-го типа (СЦА 1), ген которой клонирован в 1993 году.
- 6. Спиноцеребеллярная атаксия 1 типа (SCA 1)- тяжелое нейродегенеративное прогрессирующее заболевание с поздним возрастом манифестации, наследуется по
- 7. Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных CAG повторов в гене ATXN1, распологающемся на 6-й хромосоме.
- 8. ЭПИДЕМИОЛОГИЯ Наиболее обширный в мире кластер СЦА1 выявлен в Якутии: достоверные сведения получены о 225 больных
- 10. КЛИНИЧЕСКАЯ КАРТИНА В типичных случаях заболевание начинается на 3—4-м десятилетии жизни (возможный разброс — от 4
- 11. Во многих случаях могут наблюдаться тремор головы, глазодвигательные, бульварные и тазовые расстройства, снижение вибрационной и суставно-мышечной
- 12. ДИАГНОСТИКА Клинический, семейный анамнез КТ, МРТ ЭМГ, ЭНМГ-исследования Молекулярно-генетические исследования
- 13. ДИАГНОСТИКА На КТ/МРТ у больных СЦА1 выявляются расширение субарахноидальных пространств полушарий и червя мозжечка, истончение средней
- 14. Морфологически СЦА1 характеризуется типичной картиной оливопонтоцеребеллярной атрофии (Robitaille Y. et al., Orr H.T.): на секции отмечаются
- 15. Однако, учитывая значительное разнообразие генетических вариантов АД СЦА, редкость многих его форм, и, соответственно, недостаточную изученность
- 16. ВЫДЕЛЕНИЕ ГЕНОМНОЙ ДНК
- 17. ПЦР – МЕТОД АМПЛИФИКАЦИИ ДНК IN VITRO
- 18. РЕСТРИКЦИОННЫЙ АНАЛИЗ
- 19. МЕТОД ЭЛЕКТРОФОРЕЗА
- 22. АНАЛИЗ РЕЗУЛЬТАТОВ 1. Автоматически по компьютерной программе BioCapt подсчитывается молекулярный вес аллелей гена. 2. По формуле
- 23. ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ ФРАГМЕНТНОГО АНАЛИЗА Нормальный аллель - 27 Патологический аллель - 55
- 24. ЛЕЧЕНИЕ Этиотропной терапии на данный момент не существует. Аминоплазмаль Е L-цистеин ( Ф.А.Платонов) Мемантин Симптоматическое лечение
- 25. ЛЕЧЕНИЕ Лечение включает проведение мероприятий, направленных на двигательную и социальную реабилитацию пациентов, их адаптацию к имеющемуся
- 26. ЛЕЧЕНИЕ Единственным способом борьбы с этим заболеванием на сегодняшний день является профилактика появления новых случаев СЦА1
- 28. Скачать презентацию

Наследственные атаксии (НА) — клинически и генетически крайне гетерогенная группа наследственных
Наследственные атаксии (НА) — клинически и генетически крайне гетерогенная группа наследственных

Аутосомно-доминантные спиноцеребеллярные атаксии (АД-СЦА) характеризуются прогрессирующей атаксией, возникающей вследствие дегенерации мозжечка,
Аутосомно-доминантные спиноцеребеллярные атаксии (АД-СЦА) характеризуются прогрессирующей атаксией, возникающей вследствие дегенерации мозжечка,

Первая классификация АД-СЦА, основанная на фенотипических характеристиках, была предложена A. Harding:
Первая классификация АД-СЦА, основанная на фенотипических характеристиках, была предложена A. Harding:

Хронологически первой формой АД-СЦА стала СЦА 1-го типа (СЦА 1), ген
Хронологически первой формой АД-СЦА стала СЦА 1-го типа (СЦА 1), ген
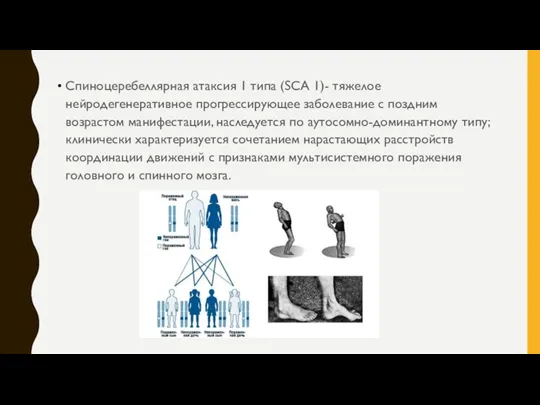
Спиноцеребеллярная атаксия 1 типа (SCA 1)- тяжелое нейродегенеративное прогрессирующее заболевание с
Спиноцеребеллярная атаксия 1 типа (SCA 1)- тяжелое нейродегенеративное прогрессирующее заболевание с

Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных CAG повторов в гене
Молекулярно-генетической причиной SCA1 является увеличение числа тринуклеотидных CAG повторов в гене

ЭПИДЕМИОЛОГИЯ
Наиболее обширный в мире кластер СЦА1 выявлен в Якутии: достоверные сведения
ЭПИДЕМИОЛОГИЯ
Наиболее обширный в мире кластер СЦА1 выявлен в Якутии: достоверные сведения


КЛИНИЧЕСКАЯ КАРТИНА
В типичных случаях заболевание начинается на 3—4-м десятилетии жизни (возможный
КЛИНИЧЕСКАЯ КАРТИНА
В типичных случаях заболевание начинается на 3—4-м десятилетии жизни (возможный

Во многих случаях могут наблюдаться тремор головы, глазодвигательные, бульварные и
Во многих случаях могут наблюдаться тремор головы, глазодвигательные, бульварные и
ДИАГНОСТИКА
Клинический, семейный анамнез
КТ, МРТ
ЭМГ, ЭНМГ-исследования
Молекулярно-генетические исследования
ДИАГНОСТИКА
Клинический, семейный анамнез
КТ, МРТ
ЭМГ, ЭНМГ-исследования
Молекулярно-генетические исследования

ДИАГНОСТИКА
На КТ/МРТ у больных СЦА1 выявляются расширение субарахноидальных пространств полушарий и
ДИАГНОСТИКА
На КТ/МРТ у больных СЦА1 выявляются расширение субарахноидальных пространств полушарий и

Морфологически СЦА1 характеризуется типичной картиной оливопонтоцеребеллярной атрофии (Robitaille Y. et al.,
Морфологически СЦА1 характеризуется типичной картиной оливопонтоцеребеллярной атрофии (Robitaille Y. et al.,

Однако, учитывая значительное разнообразие генетических вариантов АД СЦА, редкость многих его
Однако, учитывая значительное разнообразие генетических вариантов АД СЦА, редкость многих его

ВЫДЕЛЕНИЕ ГЕНОМНОЙ ДНК
ВЫДЕЛЕНИЕ ГЕНОМНОЙ ДНК
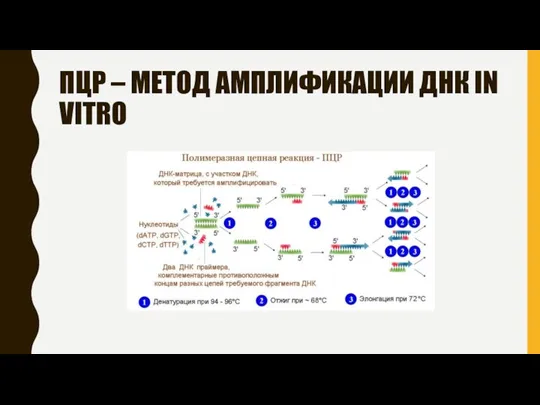
ПЦР – МЕТОД АМПЛИФИКАЦИИ ДНК IN VITRO
ПЦР – МЕТОД АМПЛИФИКАЦИИ ДНК IN VITRO
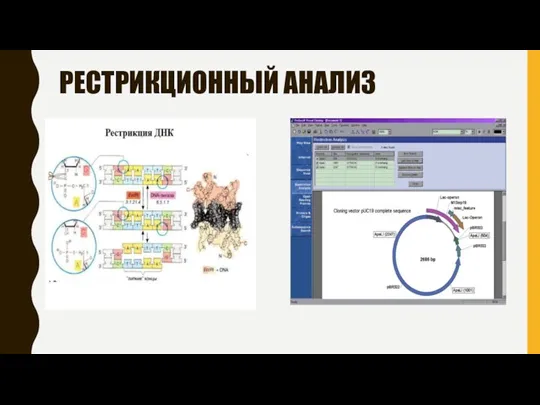
РЕСТРИКЦИОННЫЙ АНАЛИЗ
РЕСТРИКЦИОННЫЙ АНАЛИЗ

МЕТОД ЭЛЕКТРОФОРЕЗА
МЕТОД ЭЛЕКТРОФОРЕЗА

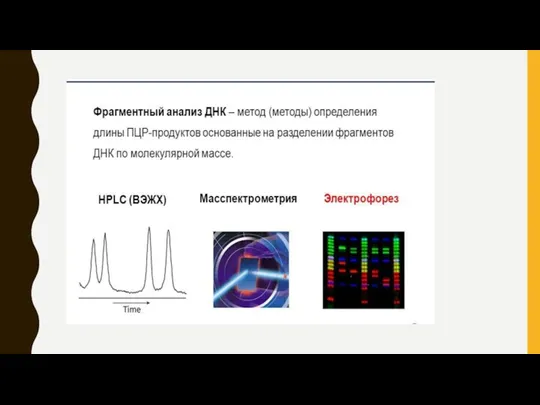

АНАЛИЗ РЕЗУЛЬТАТОВ
1. Автоматически по компьютерной программе BioCapt подсчитывается молекулярный вес аллелей
АНАЛИЗ РЕЗУЛЬТАТОВ
1. Автоматически по компьютерной программе BioCapt подсчитывается молекулярный вес аллелей
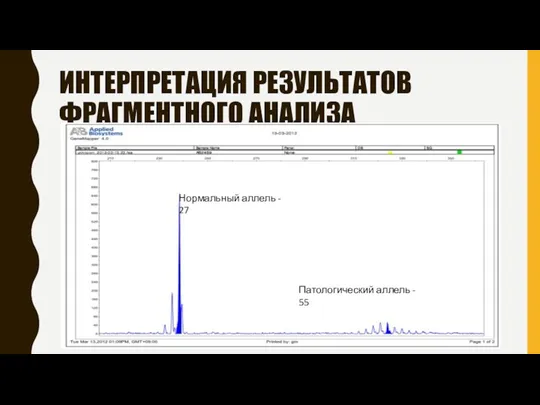
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ ФРАГМЕНТНОГО АНАЛИЗА
Нормальный аллель - 27
Патологический аллель - 55
ИНТЕРПРЕТАЦИЯ РЕЗУЛЬТАТОВ ФРАГМЕНТНОГО АНАЛИЗА
Нормальный аллель - 27
Патологический аллель - 55

ЛЕЧЕНИЕ
Этиотропной терапии на данный момент не существует.
Аминоплазмаль Е
L-цистеин ( Ф.А.Платонов)
Мемантин
Симптоматическое лечение
ЛЕЧЕНИЕ
Этиотропной терапии на данный момент не существует.
Аминоплазмаль Е
L-цистеин ( Ф.А.Платонов)
Мемантин
Симптоматическое лечение

ЛЕЧЕНИЕ
Лечение включает проведение мероприятий, направленных на двигательную и социальную реабилитацию пациентов,
ЛЕЧЕНИЕ
Лечение включает проведение мероприятий, направленных на двигательную и социальную реабилитацию пациентов,

ЛЕЧЕНИЕ
Единственным способом борьбы с этим заболеванием на сегодняшний день является профилактика
ЛЕЧЕНИЕ
Единственным способом борьбы с этим заболеванием на сегодняшний день является профилактика
 Акушериядағы зерттеу әдістері
Акушериядағы зерттеу әдістері Первая медицинская помощь при кровотечении
Первая медицинская помощь при кровотечении Жедел коронарлы синдром
Жедел коронарлы синдром Почесуха
Почесуха Общая онкология
Общая онкология Методи дослiдження плоскостопостi
Методи дослiдження плоскостопостi Причины феохромоцитомы
Причины феохромоцитомы Двигательная система
Двигательная система Клостридии - возбудители раневых инфекций
Клостридии - возбудители раневых инфекций Пластикалық хирургия
Пластикалық хирургия Меланома кожи. Диагностика, клиника и лечение
Меланома кожи. Диагностика, клиника и лечение Ожирение. Классификация ожирения
Ожирение. Классификация ожирения Эшерихии. Эшерихиозы
Эшерихии. Эшерихиозы Возрастные особенности строения уха
Возрастные особенности строения уха Патофизиология сердечно-сосудистой системы. Лекция 2
Патофизиология сердечно-сосудистой системы. Лекция 2 Вред наркотиков. Профилактика наркомании и формирование установок на ведение здорового образа жизни
Вред наркотиков. Профилактика наркомании и формирование установок на ведение здорового образа жизни Коленный сустав
Коленный сустав Приказ Минздрава РФ от 16.07.97 n 214 О контроле качества лекарственных средств, изготовляемых в аптеках
Приказ Минздрава РФ от 16.07.97 n 214 О контроле качества лекарственных средств, изготовляемых в аптеках Нәрестелердегі іріңді қабыну ауруы
Нәрестелердегі іріңді қабыну ауруы Истмико-цервикальная недостаточность (ИЦН)
Истмико-цервикальная недостаточность (ИЦН) Гаметогенез. Сперматогенез. Овогенез
Гаметогенез. Сперматогенез. Овогенез Сирингомиелия
Сирингомиелия Шаншуға арналған дәрілік препараттар
Шаншуға арналған дәрілік препараттар Поствакцинальные реакции и осложнения
Поствакцинальные реакции и осложнения Акушерский перитонит
Акушерский перитонит Неврологические осложнения остеохондроза позвоночника
Неврологические осложнения остеохондроза позвоночника Стоматологиядағы магниттік-резонанстық томография
Стоматологиядағы магниттік-резонанстық томография Физиология спинного мозга. (Лекция 7)
Физиология спинного мозга. (Лекция 7)