- Идиопатические воспалительные миопатии: полимиозит и дерматомиозит

Содержание
- 2. Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением которых является симметричная мышечная слабость
- 3. Классификация идиопатических воспалительных миопатий (модиф. Miller 1994) 1.Первичный идиопатический полимиозит (ПМ) 2.Первичный идиопатический дерматомиозит (ДМ) 3.Миозит,
- 4. Эпидемиология Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели заболеваемости составляют от 2,18
- 5. Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и гладкой мускулатуры с нарушением двигательной
- 6. Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный (идиопатический) полимиозит II. Первичный (идиопатический)
- 7. Симптоматика Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь преимущественно кожными и мышечными
- 8. Симптоматика При ПМ поражение кожи отсутствует, но уже с начала заболевания остро или постепенно развивается характерная
- 9. Симптоматика Мышечный синдром проявляется в первую очередь болями в мышцах при движении, прощупывании и даже в
- 10. Симптоматика Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода. В таких случаях развивается
- 11. Симптоматика Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже при полимиозите наблюдается поражение
- 12. Клинические признаки Повышение температуры тела Поражение кожи: эритема периорбитальный отек капилляриты отек Синдром Рейно Генерализованное поражение
- 13. Диагностические критерии ПМ/ДМ (Bohan, Peter 1975) 1. Симметричная проксимальная слабость мышц плечевого и тазового пояса, нарастающая
- 14. Лабораторно- инструментальные методы исследования Увеличение КФК, АЛТ, АСТ, ЛДГ. Аутоантитела обнаруживаются в сыворотке пациентов 50% ПМ/ДМ.
- 15. Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические фенотипы. К ним относятся анти-Мi-2,
- 16. Морфологическое исследование. Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной инфильтрации плазмой и воспалительными клетками
- 17. При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают в неизмененные мышечные волокна, экспрессирующие
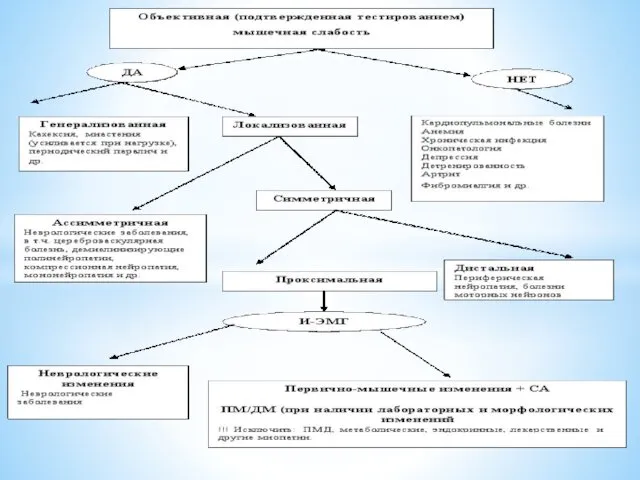
- 18. Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью (не получающих ГК):
- 20. Методы оценки мышечной силы. Мануальное тестирование силы проксимальных и аксиальных мышц проводится согласно рекомендациям IMACS и
- 21. Лечение ПМ/ДМ !!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая мышечную ткань, может
- 22. Лечение ПМ/ДМ • Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с
- 23. Лечение ПМ/ДМ • Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца. • Снижение дозы ГК
- 24. Лечение ПМ/ДМ • Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и не служит
- 25. Лечение ПМ/ДМ Потенциальные показания к подключению иммуносупрессивной терапии : • Принадлежность больных к клинико-иммунологическим подтипам ПМ/ДМ,
- 26. Лечение ПМ/ДМ • Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или плохой переносимости
- 27. Лечение ПМ/ДМ Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к другим метода лечения
- 28. Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной дозой ГК
- 30. Скачать презентацию

Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением
Идиопатические воспалительные миопатии — группа хронических заболеваний, неизвестной этиологии, основным проявлением

Классификация идиопатических воспалительных миопатий (модиф. Miller 1994)
1.Первичный идиопатический полимиозит (ПМ)
2.Первичный идиопатический
Классификация идиопатических воспалительных миопатий (модиф. Miller 1994)
1.Первичный идиопатический полимиозит (ПМ)
2.Первичный идиопатический

Эпидемиология
Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели
Эпидемиология
Распространенность и частота варьирует в различных популяциях. Согласно эпидемиологическим исследованиям, показатели

Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и
Дерматомиозит (ДМ) — системное прогрессирующее заболевание с преимущественным поражением поперечно-полосатой и

Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный
Классификация дерматомиозита (полимиозита) по А. Bohan и Y.Peter (1975) I. Первичный

Симптоматика
Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь
Симптоматика
Начало заболевания может быть острым, но чаще симптоматика развивается постепенно, характеризуясь

Симптоматика
При ПМ поражение кожи отсутствует, но уже с начала заболевания остро
Симптоматика
При ПМ поражение кожи отсутствует, но уже с начала заболевания остро

Симптоматика
Мышечный синдром проявляется в первую очередь болями в мышцах при движении,
Симптоматика
Мышечный синдром проявляется в первую очередь болями в мышцах при движении,

Симптоматика
Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода.
Симптоматика
Возможно течение полимиозита с поражением гладкой мускулатуры глотки, гортани и пищевода.

Симптоматика
Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже
Симптоматика
Суставной синдром характеризуется поражением лучезапястных суставов и мелких суставов кисти. Реже

Клинические признаки
Повышение температуры тела
Поражение кожи:
эритема
периорбитальный отек
капилляриты
отек
Синдром Рейно
Генерализованное
Клинические признаки
Повышение температуры тела
Поражение кожи:
эритема
периорбитальный отек
капилляриты
отек
Синдром Рейно
Генерализованное

Диагностические критерии ПМ/ДМ (Bohan, Peter 1975)
1. Симметричная проксимальная слабость мышц плечевого
Диагностические критерии ПМ/ДМ (Bohan, Peter 1975)
1. Симметричная проксимальная слабость мышц плечевого

Лабораторно- инструментальные методы исследования
Увеличение КФК, АЛТ, АСТ, ЛДГ.
Аутоантитела обнаруживаются в
Лабораторно- инструментальные методы исследования
Увеличение КФК, АЛТ, АСТ, ЛДГ.
Аутоантитела обнаруживаются в

Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические
Миозит-специфические антитела выявляются только при ИВМ и далее маркеруют их клинические

Морфологическое исследование.
Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной
Морфологическое исследование.
Дерматомиозит является комплемент-зависимой микроангиопатией, ведущей к разрушению капилляров, повышенной

При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают
При полимиозите наблюдаются множественные очаги воспаления, где выявляются CD8+Т-клетки, которые проникают

Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью
Алгоритм диагностического поиска и дифференциальный диагноз у пациентов с мышечной слабостью
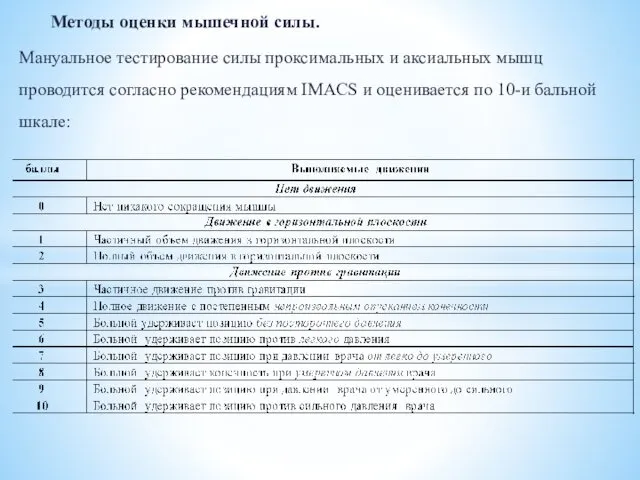
Методы оценки мышечной силы.
Мануальное тестирование силы проксимальных и аксиальных мышц
Методы оценки мышечной силы.
Мануальное тестирование силы проксимальных и аксиальных мышц

Лечение ПМ/ДМ
!!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых,
Лечение ПМ/ДМ
!!! У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых,

Лечение ПМ/ДМ
• Раннее начало терапии (в течение первых 3-х месяцев от начала
Лечение ПМ/ДМ
• Раннее начало терапии (в течение первых 3-х месяцев от начала

Лечение ПМ/ДМ
• Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
• Снижение дозы
Лечение ПМ/ДМ
• Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
• Снижение дозы

Лечение ПМ/ДМ
• Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ
Лечение ПМ/ДМ
• Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ

Лечение ПМ/ДМ
Потенциальные показания к подключению иммуносупрессивной терапии :
• Принадлежность больных к клинико-иммунологическим
Лечение ПМ/ДМ
Потенциальные показания к подключению иммуносупрессивной терапии :
• Принадлежность больных к клинико-иммунологическим

Лечение ПМ/ДМ
• Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности
Лечение ПМ/ДМ
• Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности

Лечение ПМ/ДМ
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным
Лечение ПМ/ДМ
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным

Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой
Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой
 Фото. Айсберг
Фото. Айсберг Остеоартроз (остеоартрит)
Остеоартроз (остеоартрит) Отчёт по работе Духовно-просветительского центра Глазовской епархии за 2019 год
Отчёт по работе Духовно-просветительского центра Глазовской епархии за 2019 год Storage devices
Storage devices Идеальные растворы. Законы Дальтона и Рауля
Идеальные растворы. Законы Дальтона и Рауля Исследовательский проект Красочные эксперименты.
Исследовательский проект Красочные эксперименты. Игры и задания на развитие интеллекта. 4 – 7 лет
Игры и задания на развитие интеллекта. 4 – 7 лет Трудовое право. Конвенция о правах ребенка. Конституция РФ. Трудовой кодекс РФ. Отдельные законы о труде
Трудовое право. Конвенция о правах ребенка. Конституция РФ. Трудовой кодекс РФ. Отдельные законы о труде Понятие об инфекционном процессе и инфекционных болезнях
Понятие об инфекционном процессе и инфекционных болезнях Датчики для метеостанции. Структурная и принципиальная схема разрабатываемой метеостанции. Конструкторские расчеты
Датчики для метеостанции. Структурная и принципиальная схема разрабатываемой метеостанции. Конструкторские расчеты Полеты в космос
Полеты в космос Простое оштукатуривание стен строительного склада цементным раствором и ремонт облицовки пола керамической метлахской плиткой
Простое оштукатуривание стен строительного склада цементным раствором и ремонт облицовки пола керамической метлахской плиткой Машинобудування та металообробка Харківської області
Машинобудування та металообробка Харківської області История России в первой половине ХХ века
История России в первой половине ХХ века Народная игрушка
Народная игрушка Сочинение на тему Чем мне запомнилась картина Валентина Александровича Серова Мика Морозов
Сочинение на тему Чем мне запомнилась картина Валентина Александровича Серова Мика Морозов Презентация Я-лидер.
Презентация Я-лидер. Профессия воспитатель
Профессия воспитатель Безработица в России: типы, социальна структура, особенности динамики в условиях формирования цифровой экономики
Безработица в России: типы, социальна структура, особенности динамики в условиях формирования цифровой экономики Death by PowerPoint (and how to fight it)
Death by PowerPoint (and how to fight it) Природные ресурсы земной коры
Природные ресурсы земной коры Свойства числовых неравенств
Свойства числовых неравенств Презентация русско-французского образовательного проекта Защитим окружающий мир вместе
Презентация русско-французского образовательного проекта Защитим окружающий мир вместе Удивительные доисторические животные Южной Америки
Удивительные доисторические животные Южной Америки Слоговая структура слова 4-го типа в предложениях
Слоговая структура слова 4-го типа в предложениях Характеристика, типы и виды волос и ногтей. Основы строения и физиологии кожи, волос и ногтей
Характеристика, типы и виды волос и ногтей. Основы строения и физиологии кожи, волос и ногтей Методический семинар Создание персонального сайта учителя
Методический семинар Создание персонального сайта учителя Решение задач по химии (ОГЭ, ЕГЭ, Олимпиады)
Решение задач по химии (ОГЭ, ЕГЭ, Олимпиады)