- Синдром Альпорта, болезнь тонких базальных мембран

Содержание
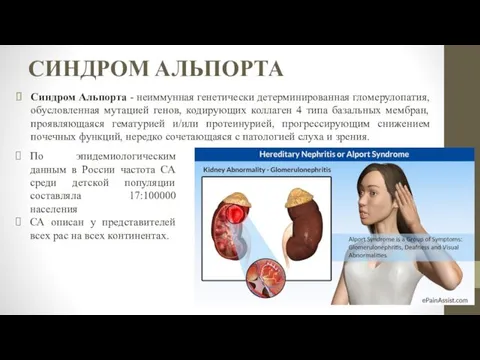
- 2. СИНДРОМ АЛЬПОРТА Синдром Альпорта - неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4 типа
- 3. ЭТИОЛОГИЯ И ПАТОГЕНЕЗ Синдром Альпорта наследуется по аутосомно-доминантному типу, частично сцеплен с полом. Больные мужчины-гетерозиготы способны
- 5. Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение и дезорганизация плотной пластины (Lamina
- 6. I вариант - клинически проявляется нефритом с гематурией, тугоухостью и поражением глаз. Течение нефрита прогрессирующее с
- 7. КЛИНИЧЕСКАЯ КАРТИНА Клиника обнаруживается на первом или (чаще) 3-5-м годах жизни ребенка. Течение наследственного нефрита может
- 8. В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного появляются: интоксикация, мышечная слабость, артериальная
- 9. Диагностика Для диагностики СА необходимо присутствие трех из пяти признаков: гематурия или летальный исход от ХПН
- 10. Фрагмент родословной семьи с синдром Альпорта
- 11. Дифференциальная диагностика С гематурической формой приобретенного гломерулонефрита. Приобретенный гломерулонефрит имеет чаще острое начало, период 2-3 нед
- 12. Лечение Антипротеинурическая терапия Первая линия (иАПФ): рамиприл в дозе 1-2 мг/м2 /сут, эналаприл в дозировке вдвое
- 13. С наступлением хронической почечной недостаточности проводится комплекс мероприятий по лечению артериальной гипертензии, анемии, электролитных и костно-минеральных
- 14. Течение и исход заболевания Течение и исход заболевания зависят от пола больных. У мальчиков рано (с
- 15. Болезнь тонких базальных мембран Доброкачественная семейная гематурия (болезнь тонких базальных мембран) - наследственное заболевание, его основной
- 18. Скачать презентацию
СИНДРОМ АЛЬПОРТА
Синдром Альпорта - неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией генов,
СИНДРОМ АЛЬПОРТА
Синдром Альпорта - неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией генов,

ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Синдром Альпорта наследуется по аутосомно-доминантному типу, частично сцеплен с
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Синдром Альпорта наследуется по аутосомно-доминантному типу, частично сцеплен с
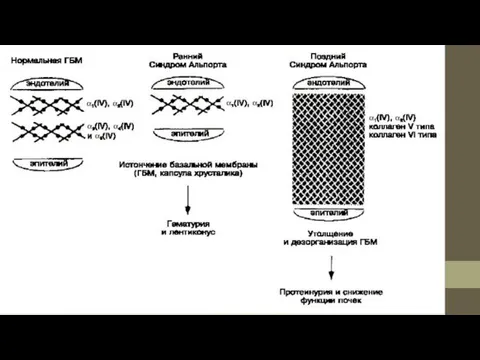

Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение
Полиморфизм ультраструктуры БМ клубочкового капилляра. Участки различной толщины, потеря трехслойности, расслоение

I вариант - клинически проявляется нефритом с гематурией, тугоухостью и поражением
I вариант - клинически проявляется нефритом с гематурией, тугоухостью и поражением

КЛИНИЧЕСКАЯ КАРТИНА
Клиника обнаруживается на первом или (чаще) 3-5-м годах жизни ребенка.
КЛИНИЧЕСКАЯ КАРТИНА
Клиника обнаруживается на первом или (чаще) 3-5-м годах жизни ребенка.

В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного
В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного

Диагностика
Для диагностики СА необходимо присутствие трех из пяти признаков:
гематурия
Диагностика
Для диагностики СА необходимо присутствие трех из пяти признаков:
гематурия
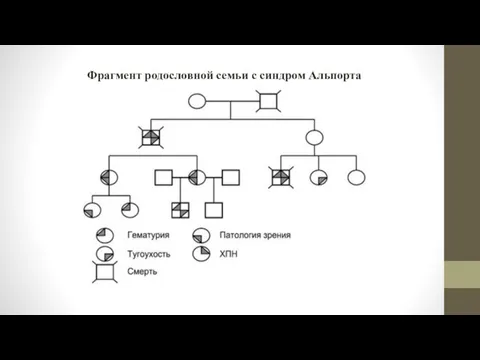
Фрагмент родословной семьи с синдром Альпорта
Фрагмент родословной семьи с синдром Альпорта

Дифференциальная диагностика
С гематурической формой приобретенного гломерулонефрита.
Приобретенный гломерулонефрит имеет чаще острое начало,
Дифференциальная диагностика
С гематурической формой приобретенного гломерулонефрита.
Приобретенный гломерулонефрит имеет чаще острое начало,

Лечение
Антипротеинурическая терапия
Первая линия (иАПФ):
рамиприл в дозе 1-2 мг/м2 /сут,
эналаприл
Лечение
Антипротеинурическая терапия
Первая линия (иАПФ):
рамиприл в дозе 1-2 мг/м2 /сут,
эналаприл

С наступлением хронической почечной недостаточности проводится комплекс мероприятий по лечению артериальной
С наступлением хронической почечной недостаточности проводится комплекс мероприятий по лечению артериальной

Течение и исход заболевания
Течение и исход заболевания зависят от пола больных.
Течение и исход заболевания
Течение и исход заболевания зависят от пола больных.

Болезнь тонких базальных мембран
Доброкачественная семейная гематурия (болезнь тонких базальных мембран) - наследственное
Болезнь тонких базальных мембран
Доброкачественная семейная гематурия (болезнь тонких базальных мембран) - наследственное
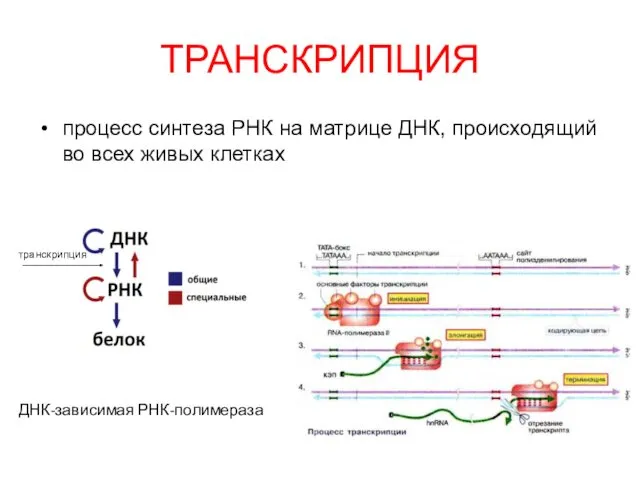 Транскрипция, процессинг
Транскрипция, процессинг Рефлексия на уроке
Рефлексия на уроке Просветительская деятельность учителя-логопеда в МДОУ Детский сад №3 Дюймовочка
Просветительская деятельность учителя-логопеда в МДОУ Детский сад №3 Дюймовочка Работа. Мехническая энергия. Кинетическая и потенциальная энергия. Закон сохранения механической энергии
Работа. Мехническая энергия. Кинетическая и потенциальная энергия. Закон сохранения механической энергии Анализ работы городского методического объединения учителей-логопедов г. Троицка за 2013-2014 учебный год
Анализ работы городского методического объединения учителей-логопедов г. Троицка за 2013-2014 учебный год Формы глагола Be в настоящем простом времени. GRAMMAR
Формы глагола Be в настоящем простом времени. GRAMMAR Геометрические приложения определенного интеграла
Геометрические приложения определенного интеграла Презентация к занятию Я люблю шоколад, а полезен ли он
Презентация к занятию Я люблю шоколад, а полезен ли он Семинар по неорганической химии: Комплексные соединения. Качественный анализ
Семинар по неорганической химии: Комплексные соединения. Качественный анализ Обережно - сказ
Обережно - сказ Компьютерная графика
Компьютерная графика Экономическое развитие России при Петре I
Экономическое развитие России при Петре I Страна вежливости и доброты
Страна вежливости и доброты Эпоха Возрождения - позднее европейское средневековье
Эпоха Возрождения - позднее европейское средневековье Места названые в честь В.Шекспира
Места названые в честь В.Шекспира Основы электроники
Основы электроники Золотодобывающая промышленность
Золотодобывающая промышленность Эксплуатация штатных образцов ВВСТ (артиллерийского вооружения)
Эксплуатация штатных образцов ВВСТ (артиллерийского вооружения) Экологическое право
Экологическое право Мировая религия пастафарианство
Мировая религия пастафарианство Muhammad and the sources
Muhammad and the sources Социальное партнёрство с родителями, как условие развития творческих способностей обучающихся
Социальное партнёрство с родителями, как условие развития творческих способностей обучающихся  Программа классного руководителя Мир начинается с меня
Программа классного руководителя Мир начинается с меня Названия чисел в записях действий. числовые выражения
Названия чисел в записях действий. числовые выражения Виды торговых помещений
Виды торговых помещений Уравнение. 5 класс
Уравнение. 5 класс Натрий (Na)
Натрий (Na) О рассмотрении паспорта проекта Модернизация ТГ-6 с заменой проточной части цилиндра среднего давления (ЦСД)
О рассмотрении паспорта проекта Модернизация ТГ-6 с заменой проточной части цилиндра среднего давления (ЦСД)