- In vivo folding. In vitro folding: spontaneously

Содержание
- 2. In vivo (in the cell): - RNA-encoded protein chain is synthesized at a ribosome. - Biosynthesis
- 3. The main obstacle for in vivo folding experiments: nascent protein is small, ribosome (+ …) is
- 4. 15N, 13C NMR: Cotranslational structure acquisition of nascent polypeptides monitored by NMR spectroscopy. Eichmann C, Preissler
- 5. 15N, 13C NMR: Monitoring cotranslational protein folding in mammalian cells at codon resolution. Han Y., David
- 6. Folding: inside or outside GroEL/ES? - OUTSIDE «Anfinsen cage»? Ellis R.J. 2003 Curr. Biol. 13:R881-3 Passive
- 7. PROTEIN CHAIN CAN FORM ITS UNIQUE 3D STRUCTURE SPONTANEOUSLY IN VITRO (Anfinsen, 1961: Nobel Prize, 1972)
- 8. ∙ In vitro (in physico-chemical experiment): Unfolded globular protein is capable of renaturation (if it is
- 9. Robert Bruce Merrifield (1921 – 2006) Nobel Prize 1988 Christian Boehmer Anfinsen, Jr. (1916 –1995) Nobel
- 10. HOW DOES PROTEIN FOLD? and even more: How CAN protein fold spontaneously? Levinthal paradox (1968): SPECIAL
- 11. “Framework model” of stepwise folding (Ptitsyn, 1973) Now: Pre-molten globule Now: Molten globule
- 12. Oleg Borisovich Ptitsyn (1929-99)
- 13. Kinetic intermediate (molten globule) in protein folding (Doldikh,…, Ptitsyn, 1984) Multi-state folding LAG
- 14. Found: metastable (“accumulating”, “directly observable”) folding intermediates. The idea was: intermediates will help to trace the
- 15. “Two-state” folding: without any observable (accumulating in experiment) intermediates The “two-state folders” fold rapidly: not only
- 16. e PROTEIN FOLDING: current picture
- 17. What to study in the “two-state” folding where there are no intermediates to single out and
- 18. “Chevron plots”: Reversible folding and unfolding even at mid-transition, where kD→N = kN→D (a) (b) N
- 19. Sir Alan Roy Fersht, 1943 Protein engineering Folding nucleus
- 20. Folding nucleus: Site-directed mutations show residues belonging and not-belonging to the “nucleus”, the native-like part of
- 21. Folding nucleus in CheY protein (Lopez-Hernandes & Serrano, 1996) In nucleus Outside “difficult” Folding nucleus
- 22. David E. Shaw “D. E. Shaw Research” US$ 3.5 billion Supercomputer “Anton” “Hot point” in protein
- 23. phase separation
- 24. “A priory” computed 3D folds of small proteins
- 25. BUT: unfolding enthalpies are predicted VERY BADLY! S. Piana, J.L. Klepeis, D.E Shaw Assessing the accuracy
- 26. Back to Levinthal paradox Native protein structure reversibly refolds from various starts, i.e., it is thermodynamically
- 27. Is “Levinthal paradox” a paradox at all? L-dimensional “Golf course”
- 28. Zwanzig, 1992; Bicout & Szabo, 2000 Is “Levinthal paradox” a paradox at all? …any tilt of
- 29. Sly simplicity of a “folding funnel” (without phase separation) - NO simultaneous explanation to (I) “all-or-none”
- 30. Phillips (1965) hypothesis: folding nucleus is formed by the N-end of the nascent protein chain, and
- 31. Sly simplicity of hierarchic folding as applied to resolve the Levinthal paradox Folding intermediates must become
- 32. 1-st order phase transition: rate of nucleation Crystallization, classic theory n ______________________________________ CONSECUTIVE REACTIONS: TRANSITION TIME
- 33. 1-st order phase transition: rate of nucleation Δμ≈-ΔT⋅(Hm /Tm) B~Hm ≈ (Tm/ΔT)3 ALL → ∞ at
- 34. Let us consider sequential folding (or unfolding) of a chain that has a large energy gap
- 35. L 1 ns ΔF #/RT ~ (1/2 ÷ 3/2) L2/3 micro loops Any stable fold is
- 36. Nucleus: not as small, it comprises 30-60% of the protein
- 37. ↓ ↓ Corr. = 0.7 loops At mid-transition intermediates do not matter…
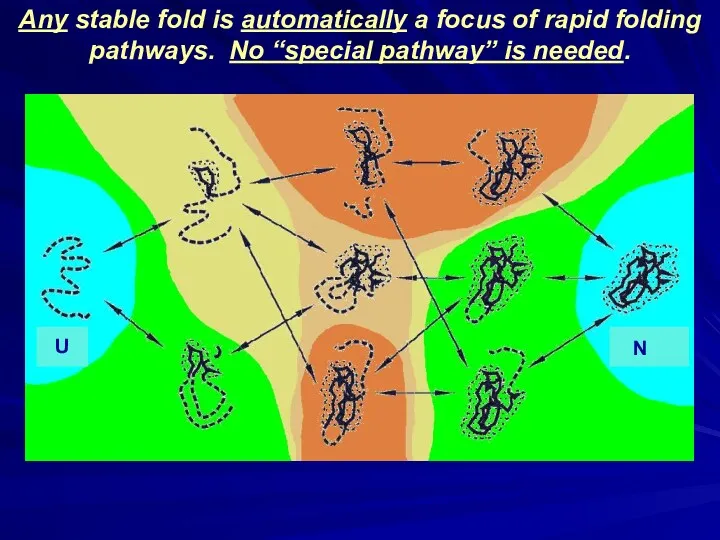
- 38. ↓ ↓ ↓ ΔFN ↓ ↓ ΔFN ↓ Any stable fold is automatically a focus of
- 39. α-helices decrease effective chain length. THIS HELPS TO FOLD! Corr. = 0.84 α-HELICES ARE PREDICTED FROM
- 40. When globules become more stable than U: a b a b ↑ GAP ⏐ ↓ 1)
- 41. Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session 77, 2003] Garbuzynskiy, Ivankov, Bogatyreva,
- 42. Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session 77, 2003] Garbuzynskiy, Ivankov, Bogatyreva,
- 45. Скачать презентацию

In vivo (in the cell):
- RNA-encoded protein chain is
In vivo (in the cell):
- RNA-encoded protein chain is
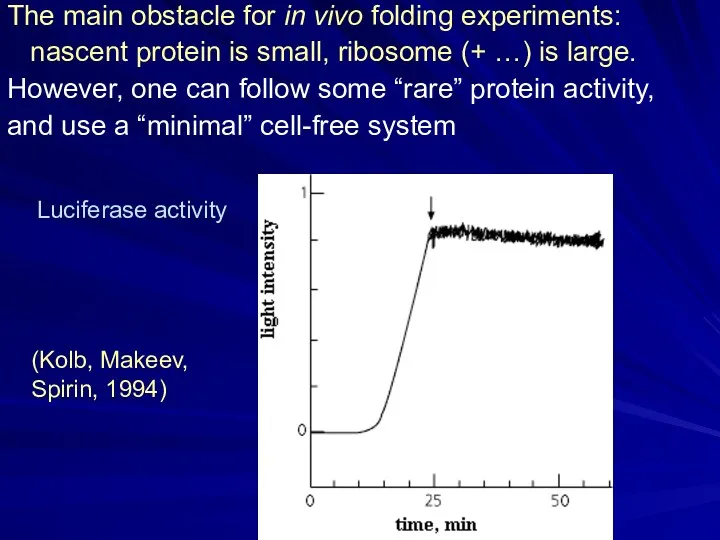
The main obstacle for in vivo folding experiments:
nascent protein is
The main obstacle for in vivo folding experiments:
nascent protein is
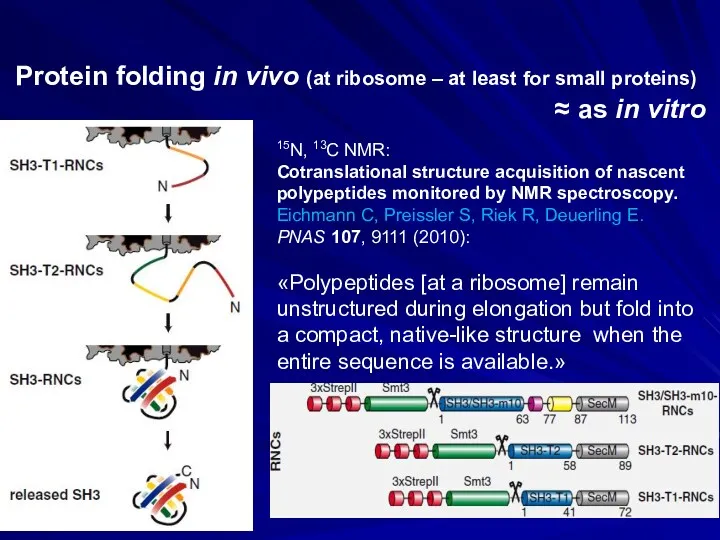
15N, 13C NMR:
Cotranslational structure acquisition of nascent polypeptides monitored by NMR
15N, 13C NMR:
Cotranslational structure acquisition of nascent polypeptides monitored by NMR

15N, 13C NMR:
Monitoring cotranslational protein folding in
mammalian cells at codon resolution.
Han
15N, 13C NMR:
Monitoring cotranslational protein folding in
mammalian cells at codon resolution.
Han
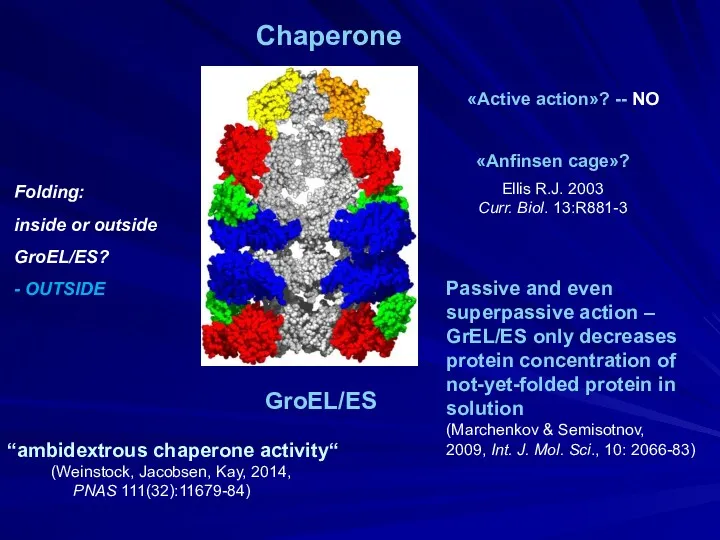
Folding:
inside or outside
GroEL/ES?
- OUTSIDE
«Anfinsen cage»?
Ellis R.J. 2003
Curr. Biol. 13:R881-3
Passive
Folding:
inside or outside
GroEL/ES?
- OUTSIDE
«Anfinsen cage»?
Ellis R.J. 2003
Curr. Biol. 13:R881-3
Passive
PROTEIN CHAIN
CAN FORM ITS UNIQUE 3D STRUCTURE
SPONTANEOUSLY IN VITRO
(Anfinsen, 1961:
PROTEIN CHAIN
CAN FORM ITS UNIQUE 3D STRUCTURE
SPONTANEOUSLY IN VITRO
(Anfinsen, 1961:

∙ In vitro (in physico-chemical experiment):
Unfolded globular protein is capable of
∙ In vitro (in physico-chemical experiment):
Unfolded globular protein is capable of

Robert Bruce
Merrifield
(1921 – 2006)
Nobel Prize 1988
Christian Boehmer
Anfinsen, Jr.
(1916
Robert Bruce
Merrifield
(1921 – 2006)
Nobel Prize 1988
Christian Boehmer
Anfinsen, Jr.
(1916
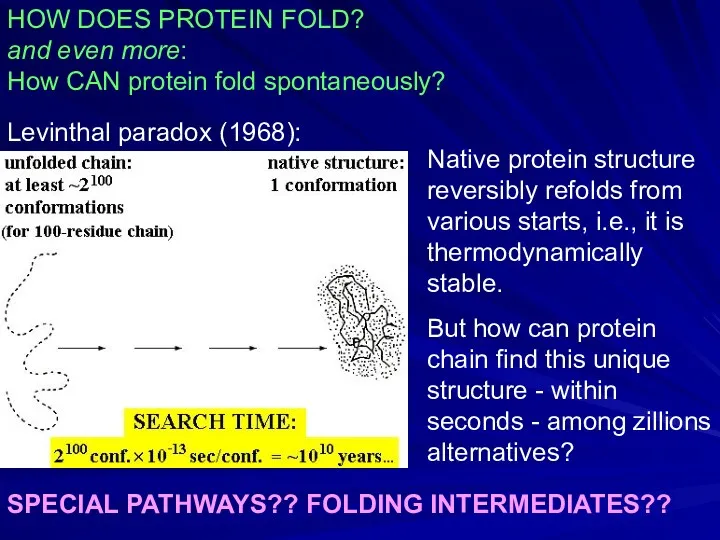
HOW DOES PROTEIN FOLD?
and even more:
How CAN protein fold spontaneously?
Levinthal paradox
HOW DOES PROTEIN FOLD?
and even more:
How CAN protein fold spontaneously?
Levinthal paradox
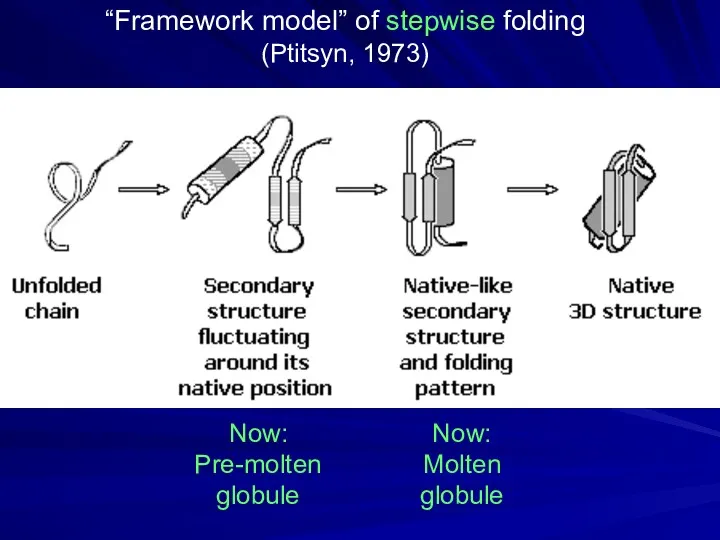
“Framework model” of stepwise folding
(Ptitsyn, 1973)
Now:
Pre-molten globule
Now: Molten globule
“Framework model” of stepwise folding
(Ptitsyn, 1973)
Now:
Pre-molten globule
Now: Molten globule

Oleg Borisovich
Ptitsyn
(1929-99)
Oleg Borisovich
Ptitsyn
(1929-99)
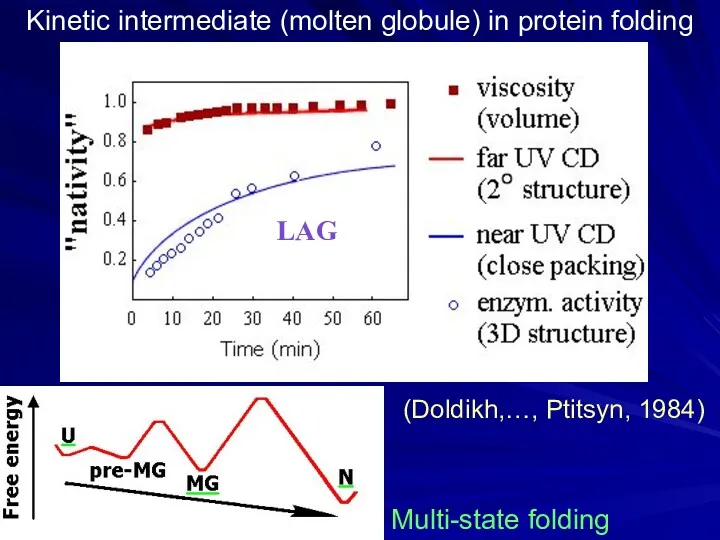
Kinetic intermediate (molten globule) in protein folding
(Doldikh,…, Ptitsyn, 1984)
Multi-state folding
LAG
Kinetic intermediate (molten globule) in protein folding
(Doldikh,…, Ptitsyn, 1984)
Multi-state folding
LAG

Found: metastable (“accumulating”, “directly observable”)
folding intermediates.
The idea was: intermediates
Found: metastable (“accumulating”, “directly observable”)
folding intermediates.
The idea was: intermediates
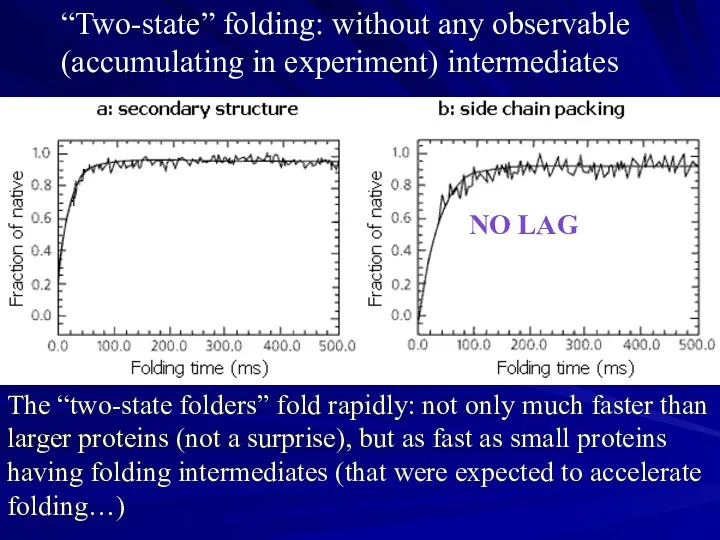
“Two-state” folding: without any observable (accumulating in experiment) intermediates
The “two-state folders”
“Two-state” folding: without any observable (accumulating in experiment) intermediates
The “two-state folders”
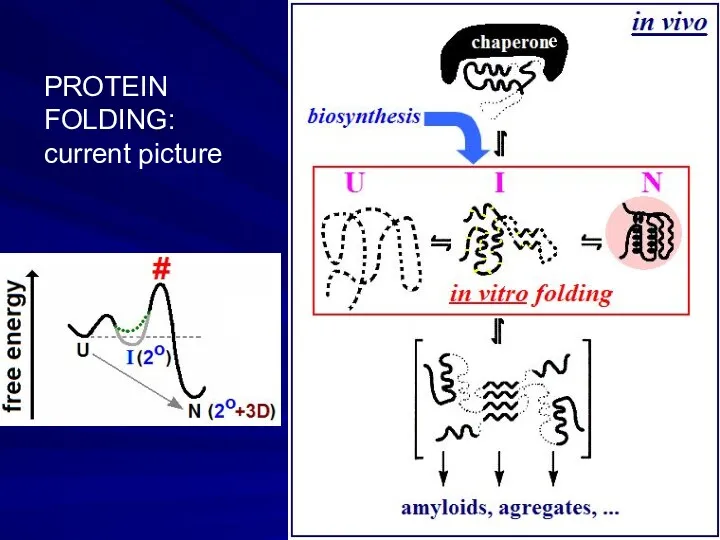
e
PROTEIN
FOLDING:
current picture
e
PROTEIN
FOLDING:
current picture

What to study in the “two-state” folding where there are no
What to study in the “two-state” folding where there are no
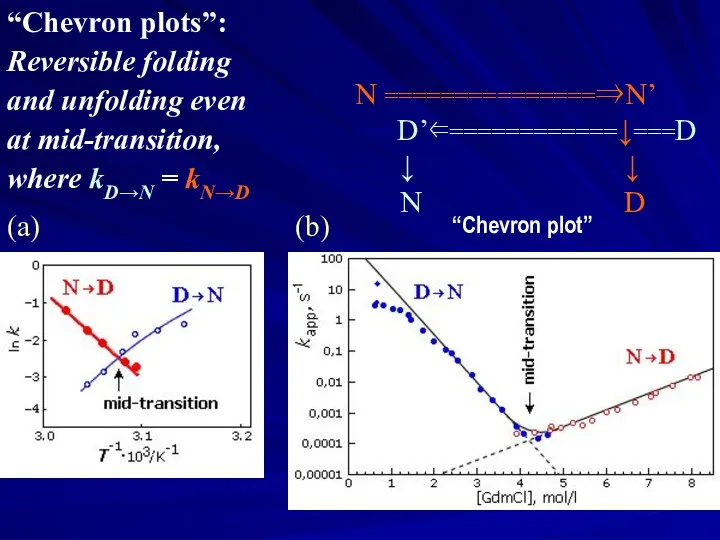
“Chevron plots”:
Reversible folding
and unfolding even
at mid-transition,
where kD→N =
“Chevron plots”:
Reversible folding
and unfolding even
at mid-transition,
where kD→N =

Sir Alan Roy Fersht, 1943
Protein engineering
Folding nucleus
Sir Alan Roy Fersht, 1943
Protein engineering
Folding nucleus
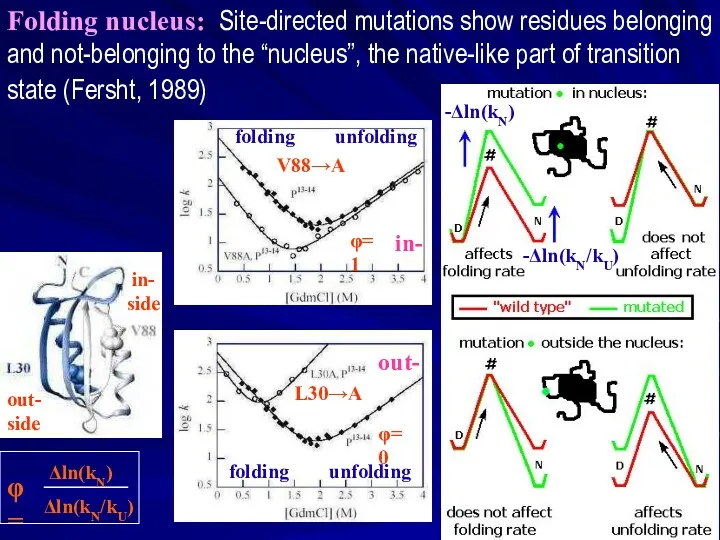
Folding nucleus: Site-directed mutations show residues belonging and not-belonging to the
Folding nucleus: Site-directed mutations show residues belonging and not-belonging to the
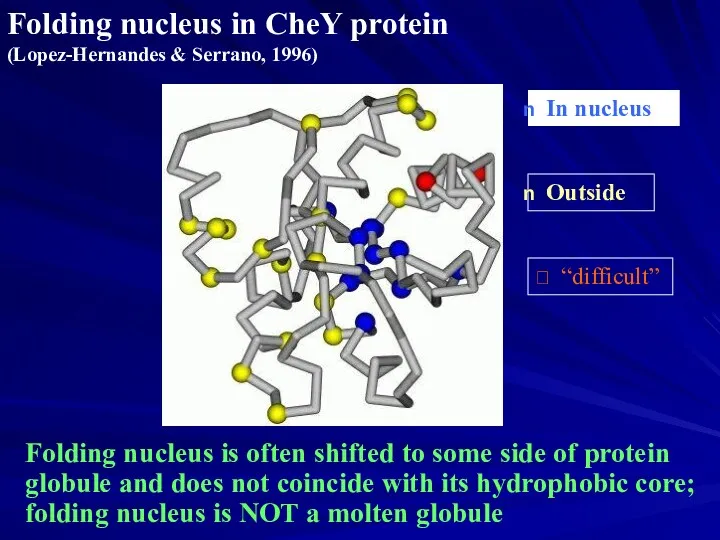
Folding nucleus in CheY protein
(Lopez-Hernandes & Serrano, 1996)
In nucleus
Folding nucleus in CheY protein
(Lopez-Hernandes & Serrano, 1996)
In nucleus

David E. Shaw
“D. E. Shaw Research”
US$ 3.5 billion
Supercomputer “Anton”
“Hot point” in
David E. Shaw
“D. E. Shaw Research”
US$ 3.5 billion
Supercomputer “Anton”
“Hot point” in
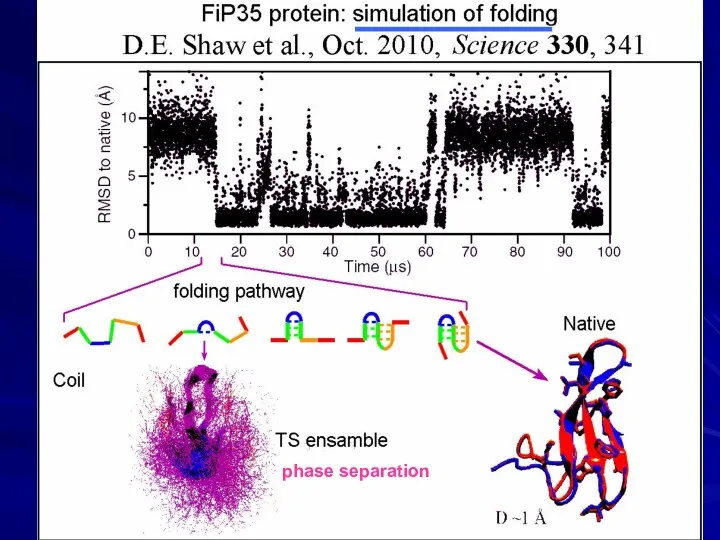
phase separation
phase separation

“A priory” computed 3D folds of small proteins
“A priory” computed 3D folds of small proteins
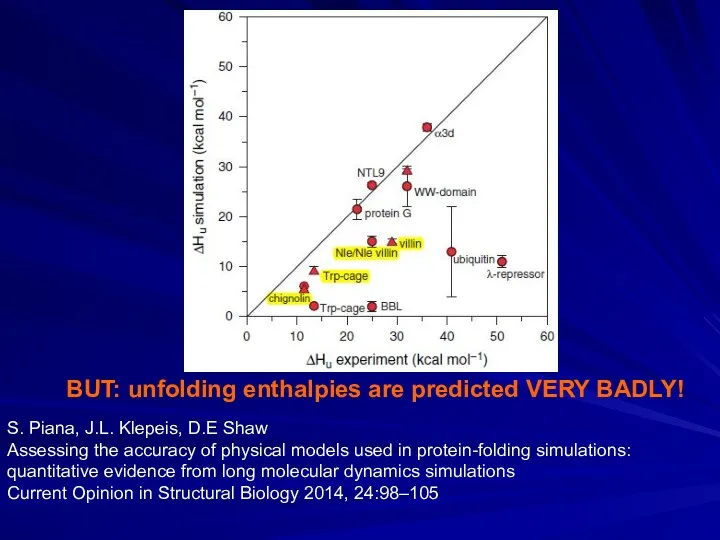
BUT: unfolding enthalpies are predicted VERY BADLY!
S. Piana, J.L. Klepeis,
BUT: unfolding enthalpies are predicted VERY BADLY!
S. Piana, J.L. Klepeis,
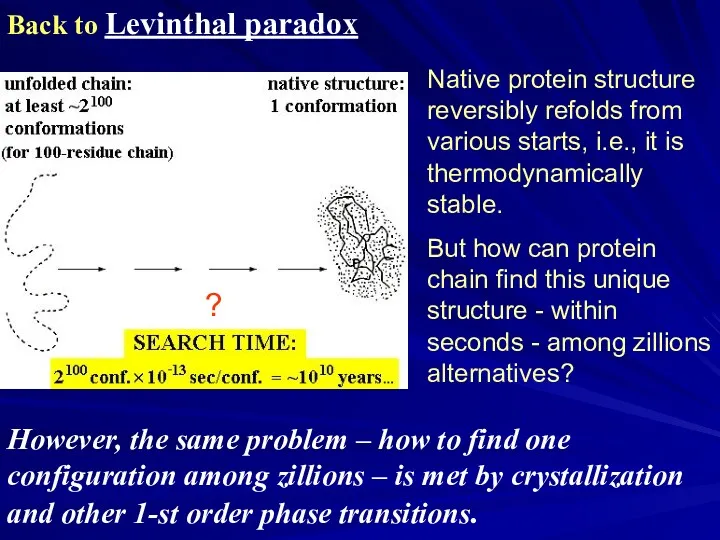
Back to Levinthal paradox
Native protein structure reversibly refolds from various starts,
Back to Levinthal paradox
Native protein structure reversibly refolds from various starts,
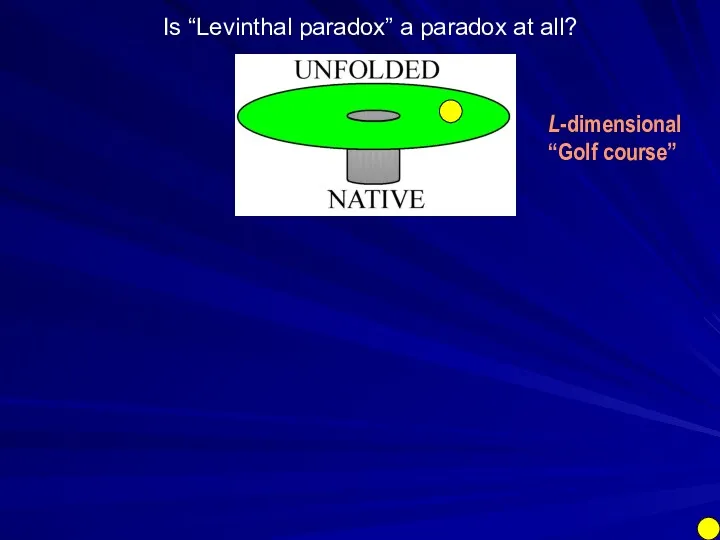
Is “Levinthal paradox” a paradox at all?
L-dimensional
“Golf course”
Is “Levinthal paradox” a paradox at all?
L-dimensional
“Golf course”
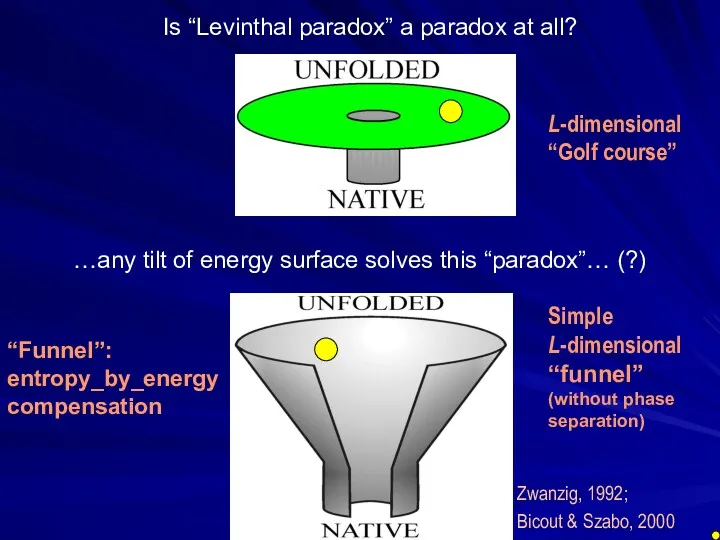
Zwanzig, 1992;
Bicout & Szabo, 2000
Is “Levinthal paradox” a paradox at
Zwanzig, 1992;
Bicout & Szabo, 2000
Is “Levinthal paradox” a paradox at
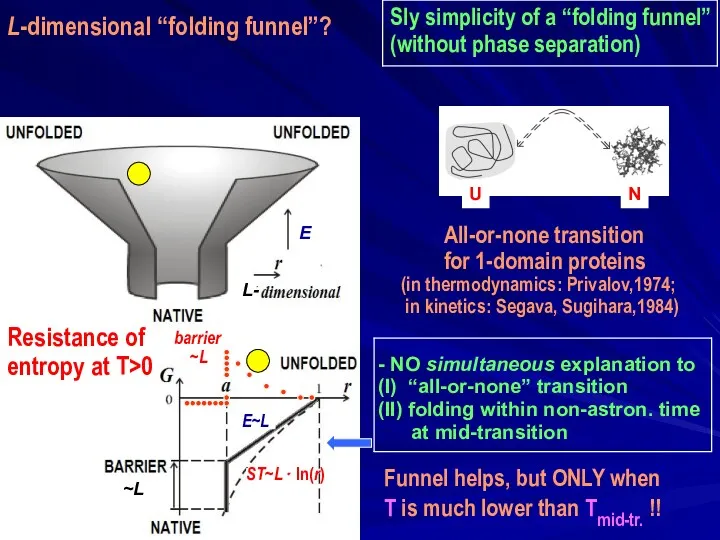
Sly simplicity of a “folding funnel”
(without phase separation)
- NO simultaneous
Sly simplicity of a “folding funnel”
(without phase separation)
- NO simultaneous

Phillips (1965) hypothesis:
folding nucleus is formed by the N-end of
Phillips (1965) hypothesis:
folding nucleus is formed by the N-end of
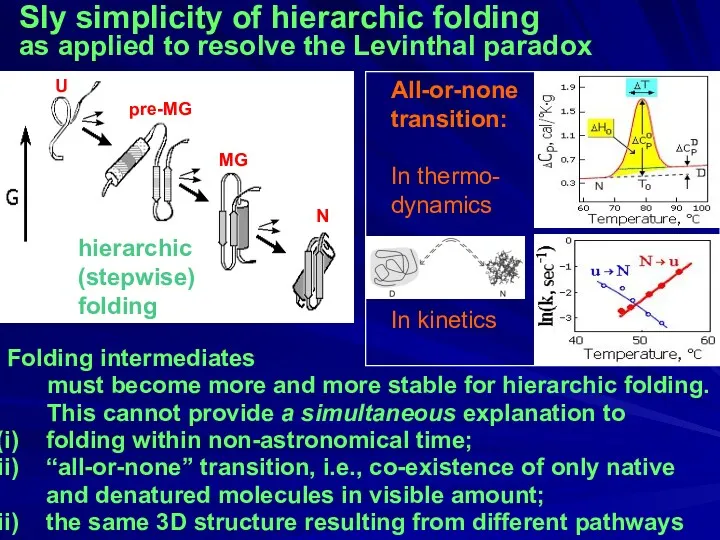
Sly simplicity of hierarchic folding
as applied to resolve the Levinthal
Sly simplicity of hierarchic folding
as applied to resolve the Levinthal

1-st order phase transition:
rate of nucleation
Crystallization, classic theory
n
______________________________________
CONSECUTIVE REACTIONS:
TRANSITION TIME ≅
1-st order phase transition:
rate of nucleation
Crystallization, classic theory
n
______________________________________
CONSECUTIVE REACTIONS:
TRANSITION TIME ≅

1-st order phase transition:
rate of nucleation
Δμ≈-ΔT⋅(Hm /Tm) B~Hm
≈ (Tm/ΔT)3
ALL → ∞
1-st order phase transition:
rate of nucleation
Δμ≈-ΔT⋅(Hm /Tm) B~Hm
≈ (Tm/ΔT)3
ALL → ∞

Let us consider sequential folding (or unfolding) of a chain that
Let us consider sequential folding (or unfolding) of a chain that
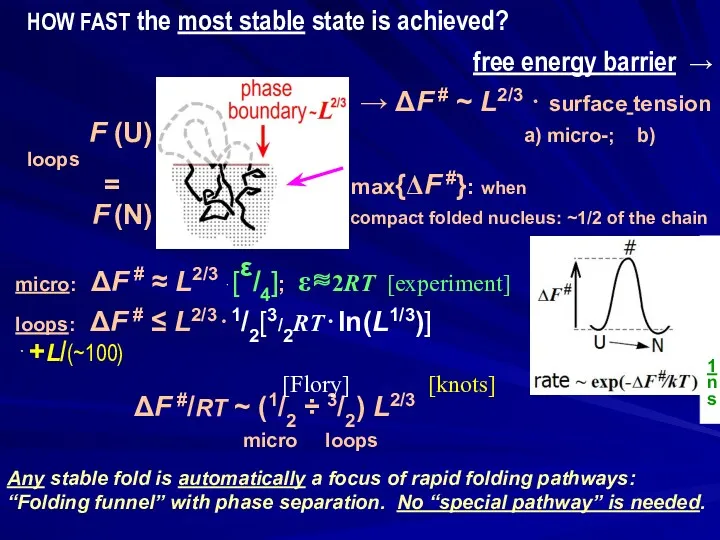
L
1
ns
ΔF #/RT ~ (1/2 ÷ 3/2) L2/3
micro loops
Any stable
L
1
ns
ΔF #/RT ~ (1/2 ÷ 3/2) L2/3
micro loops
Any stable
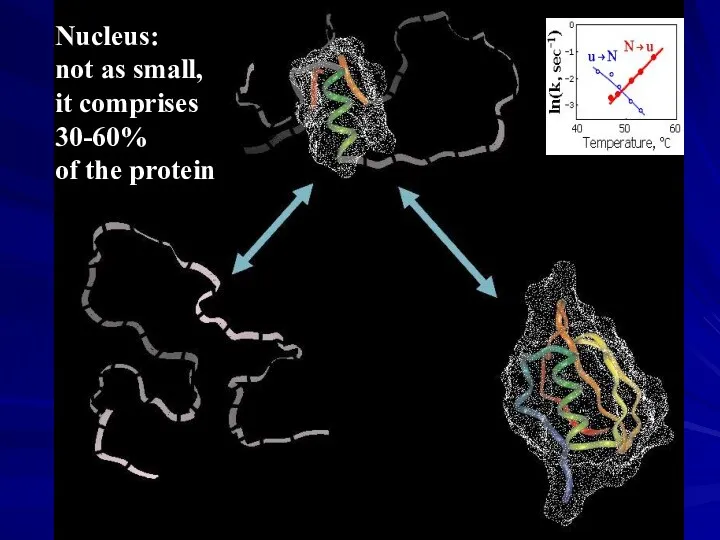
Nucleus:
not as small,
it comprises
30-60%
of the protein
Nucleus:
not as small,
it comprises
30-60%
of the protein
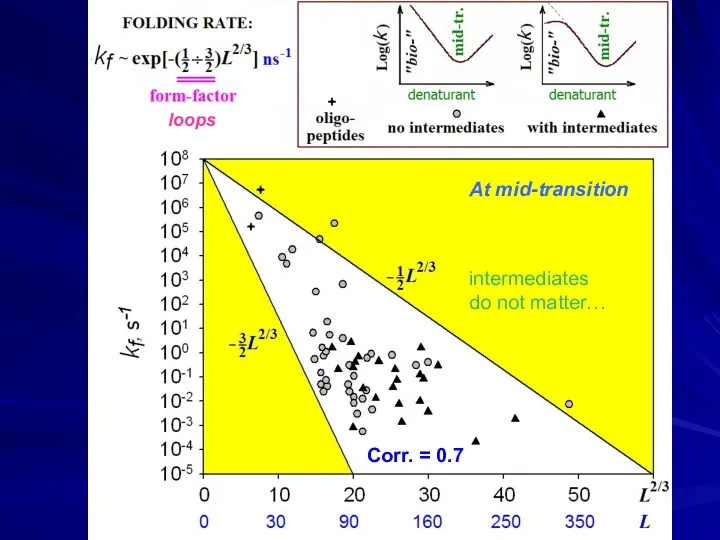
↓ ↓
Corr. = 0.7
loops
At mid-transition
intermediates
do not matter…
↓ ↓
Corr. = 0.7
loops
At mid-transition
intermediates
do not matter…
↓
↓
↓
ΔFN ↓
↓
ΔFN ↓
Any stable
↓
↓
↓
ΔFN ↓
↓
ΔFN ↓
Any stable
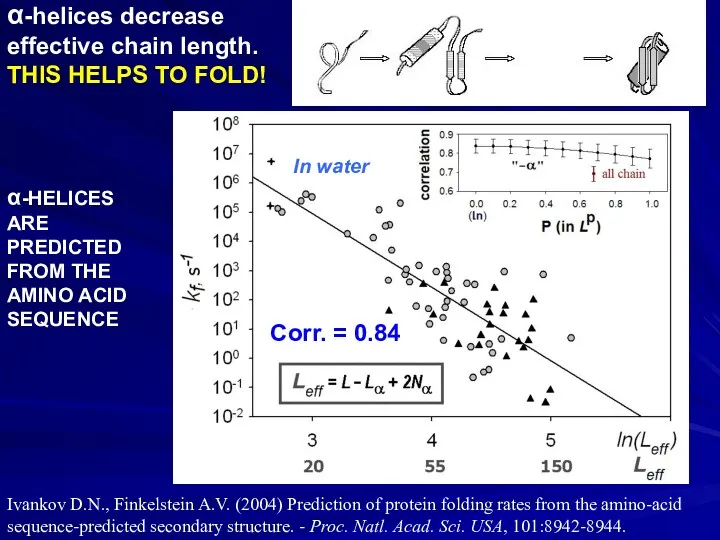
α-helices decrease
effective chain length. THIS HELPS TO FOLD!
Corr. = 0.84
α-HELICES
ARE
PREDICTED
FROM THE
AMINO
α-helices decrease
effective chain length. THIS HELPS TO FOLD!
Corr. = 0.84
α-HELICES
ARE
PREDICTED
FROM THE
AMINO
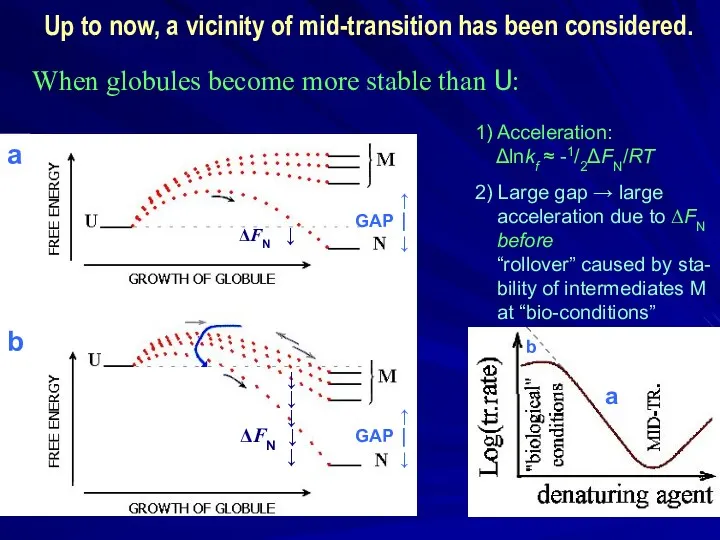
When globules become more stable than U:
a
b
a
b
↑
GAP ⏐
↓
1) Acceleration:
When globules become more stable than U:
a
b
a
b
↑
GAP ⏐
↓
1) Acceleration:
![Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches,](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/249784/slide-40.jpg)
Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session
Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session
![Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches,](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/249784/slide-41.jpg)
Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session
Finkelstein, Badretdinov; Folding & Design, 1997, 1998]. Finkelstein; Les Houches, Session
 Гидравлика
Гидравлика урок-игра по физике по теме Плотность
урок-игра по физике по теме Плотность Анализ технического паспорта асинхронного электродвигателя
Анализ технического паспорта асинхронного электродвигателя Мегаконструкції. Найбільший в світі екскаватор
Мегаконструкції. Найбільший в світі екскаватор Современные автомобили и двигатели
Современные автомобили и двигатели Внедрение ФГОС общего образования второго поколения по физике
Внедрение ФГОС общего образования второго поколения по физике Технология мини-исследования на уроках физики
Технология мини-исследования на уроках физики спектры
спектры Bbs wheels range 1999 ilustrated catalog
Bbs wheels range 1999 ilustrated catalog Законам механики подчиняются движения всех окружающих нас тел
Законам механики подчиняются движения всех окружающих нас тел Конкурс учителей Есть идея!
Конкурс учителей Есть идея! Олимпийские игры и физика
Олимпийские игры и физика Памятка по неисправностям автосцепного устройства при техническом обслуживании
Памятка по неисправностям автосцепного устройства при техническом обслуживании Урок физики в 10 классе Газовые законы
Урок физики в 10 классе Газовые законы Звуковые волны
Звуковые волны Элементарные частицы. Микромир
Элементарные частицы. Микромир Идеальный газ. Основное уравнение МКТ идеального газа
Идеальный газ. Основное уравнение МКТ идеального газа Нормирование точности поверхностей деталей машин по взаимному расположению, обозначения их на чертежах
Нормирование точности поверхностей деталей машин по взаимному расположению, обозначения их на чертежах презентация по теме Сила трения.
презентация по теме Сила трения. Стискання газів. Рівняння Менделєєва-Клапейрона
Стискання газів. Рівняння Менделєєва-Клапейрона Axles and shafts
Axles and shafts дополнение к уроку физики Испарение
дополнение к уроку физики Испарение Изобретени радио А.С. Поповым
Изобретени радио А.С. Поповым Гармонические колебания и их характеристики
Гармонические колебания и их характеристики Цепные передачи
Цепные передачи Mechanism
Mechanism Classical angular momentum and magnetic dipole moment
Classical angular momentum and magnetic dipole moment Кинематика вращательного движения
Кинематика вращательного движения