- Реакция Бэйлиса Хиллмана-Мориты

Содержание
- 2. К истории В патенте от 1972 года было описано странное, на первый взгляд, превращение: внедрение альдегидного
- 3. История Взаимодействие альдегидов с непредельными соединениями в присутствии третичных фосфинов было описано японским химиком Морита с
- 4. Роль Мориты и кто был Хиллман The Baylis–Hillman reaction is a carbon-carbon bond forming reaction between
- 5. Механизм реакции Механизм реакции предполагает обратимое присоединение амина (фосфина) по двойной связи с образованием цвиттериона 2.
- 6. Ещё один механизм Третичный амин присоединяется к активированному алкену 2, с образованием цвиттер-ионного енолята 4. Таким
- 7. Хиральные катализаторы При использовании хиральных катализаторов, становится возможным получение оптически активных продуктов.[3]
- 8. О роли квантовой химии в установлении механизмов Химики часто прибегают к методам компьютерного моделирования для того,
- 9. Авторы работы [J. Am. Chem. Soc. 2015, DOI: 10.1021/ja5111392] говорят о том, что многие механизмы многостадийных
- 10. Расчет и эксперимент На диаграмме представлены энергии интермедиатов и переходных состояний реакции Мориты-Бэйлиса-Хиллмана, определенных в ряде
- 11. Исследование, проведенное Эриком Плата (R. Erik Plata) и Даниэлом Синглтоном (Daniel A. Singleton), было посвящено реакции
- 12. Специалист по теоретической химии Кендалл Хук (Kendall N. Houk) из Университета Калифорнии (Лос-Анжелес) отмечает, что статья
- 13. Выводом статьи является то, что «теоретические исследования сложных многомолекулярных полярных реакций в растворах следует проводить и
- 14. R. l Robiette,† V. K Aggarwal,* and J. N. Harvey* Mechanism of the Morita-Baylis-Hillman Reaction: A
- 15. Что же делать? Scheme 26. Mechanism of Baylis–Hillman reaction of methyl acrylate (2a) and aldehydes 4a,b
- 17. Литература K. Morita, Z. Suzuki, H. Hirose. A Tertiary Phosphine-catalyzed Reaction of Acrylic Compounds with Aldehydes.
- 18. Интернет-ресурсы http://www.ioc.ac.ru/lib_journals/index.html 2. Проект Научная электронная библиотека (www.elibrary.ru). 3. Доступ к полным текстам журналов через электронную
- 19. Механизм реакции Hoffmann first proposed a mechanism for the MBH reaction.[9] The first reaction step involves
- 20. However, this initial mechanistic proposal had been criticized because of several points. The rate of MBH
- 22. While McQuade's and Aggarwal's studies are receiving much attention recently, there are a number of issues
- 23. MBH reaction has several advantages as a useful synthetic method: 1) It is an atom-economic coupling
- 24. Implications on Asymmetric Catalysis[edit] Nonetheless, the Aggarwal model shed light on the asymmetric catalysis of the
- 26. Sila-MBH reaction Sila-MBH reaction is a MBH variant that couples α-silylated vinyl aryl ketones with aldehydes
- 27. Rauhut-Currier reaction Rauhut-Currier reaction is a reaction of activated alkene and a Michael acceptor, not an
- 28. Tandem reaction / Multicomponent one-pot reaction Multicomponent reaction strategy is attractive for its atom-economical virtue. MBH
- 29. In addition, activated acetylenes can be added to electrophiles following a Michael addition. Trimethylsilyl iodide as
- 30. Asymmetric MBH reaction Oppolzer’s sultam can be used as a chiral auxiliary for an asymmetric MBH
- 31. Chiral allenes and imines can be employed for an asymmetric DABCO-catalyzed aza-MBH reaction.[26] Optically active 10-phenylsulfonylisobornyl
- 32. Chiral Lewis base catalyst Chiral tertiary amine catalysts are employed for enantioselective MBH reactions. β-ICD, a
- 33. Simple diamine molecules can also be employed as MBH catalysts. Methyl vinyl ketone and various substituted
- 34. Chiral phosphine MBH catalysts Chiral phosphine MBH catalysts often contain Bronsted acid moiety in their chiral
- 35. In addition, a distinct class of chiral phosphine-squaramide Квадратная кислота molecules could effectively catalyze an intramolecular
- 36. Chiral Lewis acid catalyst Chiral Lewis acid catalysts have been given interests as they could activate
- 37. Complex of metal salt and chiral ligand is a viable strategy, too. La(OTf)3 and camphor-derived chiral
- 38. Chiral Bronsted acid cocatalyst A variety of chiral thiourea catalysts are under investigation for asymmetric MBH
- 39. While simple thiourea requires a nucleophilic catalyst in conjunction, bifunctional catalysts such as phosphine-thioureas can be
- 40. Applications in Organic Synthesis
- 42. Скачать презентацию

К истории
В патенте от 1972 года было описано странное, на первый
К истории
В патенте от 1972 года было описано странное, на первый

История
Взаимодействие альдегидов с непредельными соединениями в присутствии третичных фосфинов было описано
История
Взаимодействие альдегидов с непредельными соединениями в присутствии третичных фосфинов было описано

Роль Мориты и кто был Хиллман
The Baylis–Hillman reaction is a carbon-carbon
Роль Мориты и кто был Хиллман
The Baylis–Hillman reaction is a carbon-carbon
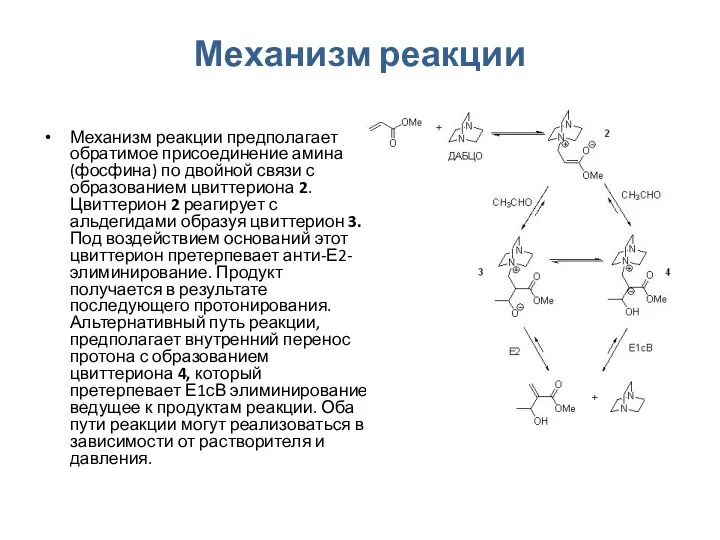
Механизм реакции
Механизм реакции предполагает обратимое присоединение амина (фосфина) по двойной связи
Механизм реакции
Механизм реакции предполагает обратимое присоединение амина (фосфина) по двойной связи

Ещё один механизм
Третичный амин присоединяется к активированному алкену 2, с
Ещё один механизм
Третичный амин присоединяется к активированному алкену 2, с
![Хиральные катализаторы При использовании хиральных катализаторов, становится возможным получение оптически активных продуктов.[3]](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/397906/slide-6.jpg)
Хиральные катализаторы
При использовании хиральных катализаторов, становится возможным получение оптически активных продуктов.[3]
Хиральные катализаторы
При использовании хиральных катализаторов, становится возможным получение оптически активных продуктов.[3]

О роли квантовой химии в установлении механизмов
Химики часто прибегают к методам
О роли квантовой химии в установлении механизмов
Химики часто прибегают к методам
![Авторы работы [J. Am. Chem. Soc. 2015, DOI: 10.1021/ja5111392] говорят](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/397906/slide-8.jpg)
Авторы работы [J. Am. Chem. Soc. 2015, DOI: 10.1021/ja5111392] говорят о
Авторы работы [J. Am. Chem. Soc. 2015, DOI: 10.1021/ja5111392] говорят о
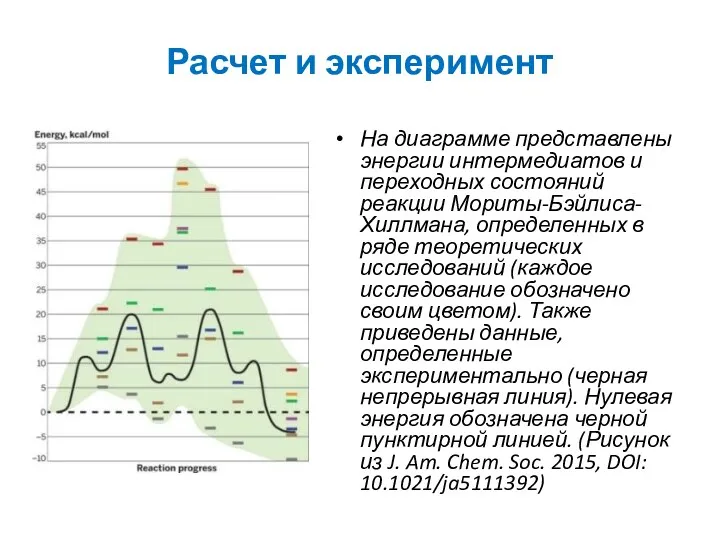
Расчет и эксперимент
На диаграмме представлены энергии интермедиатов и переходных состояний реакции
Расчет и эксперимент
На диаграмме представлены энергии интермедиатов и переходных состояний реакции

Исследование, проведенное Эриком Плата (R. Erik Plata) и Даниэлом Синглтоном (Daniel
Исследование, проведенное Эриком Плата (R. Erik Plata) и Даниэлом Синглтоном (Daniel

Специалист по теоретической химии Кендалл Хук (Kendall N. Houk) из Университета
Специалист по теоретической химии Кендалл Хук (Kendall N. Houk) из Университета

Выводом статьи является то, что «теоретические исследования сложных многомолекулярных полярных реакций
Выводом статьи является то, что «теоретические исследования сложных многомолекулярных полярных реакций
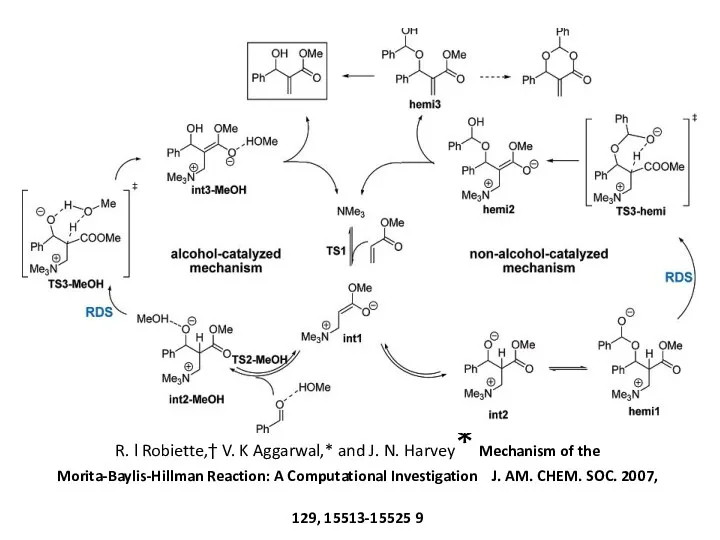
R. l Robiette,† V. K Aggarwal,* and J. N. Harvey* Mechanism
R. l Robiette,† V. K Aggarwal,* and J. N. Harvey* Mechanism
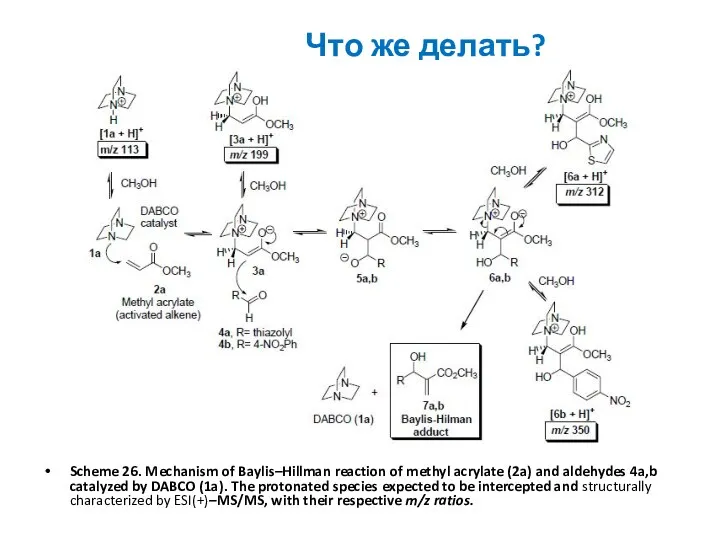
Что же делать?
Scheme 26. Mechanism of Baylis–Hillman reaction of methyl
Что же делать?
Scheme 26. Mechanism of Baylis–Hillman reaction of methyl


Литература
K. Morita, Z. Suzuki, H. Hirose. A Tertiary Phosphine-catalyzed Reaction of
Литература
K. Morita, Z. Suzuki, H. Hirose. A Tertiary Phosphine-catalyzed Reaction of

Интернет-ресурсы
http://www.ioc.ac.ru/lib_journals/index.html
2. Проект Научная электронная библиотека (www.elibrary.ru).
3. Доступ к
Интернет-ресурсы
http://www.ioc.ac.ru/lib_journals/index.html
2. Проект Научная электронная библиотека (www.elibrary.ru).
3. Доступ к
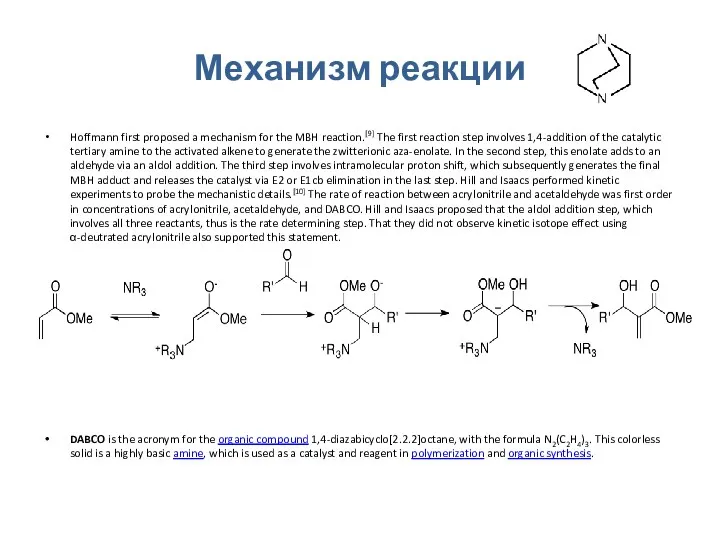
Механизм реакции
Hoffmann first proposed a mechanism for the MBH reaction.[9] The
Механизм реакции
Hoffmann first proposed a mechanism for the MBH reaction.[9] The

However, this initial mechanistic proposal had been criticized because of several
However, this initial mechanistic proposal had been criticized because of several


While McQuade's and Aggarwal's studies are receiving much attention recently, there
While McQuade's and Aggarwal's studies are receiving much attention recently, there

MBH reaction has several advantages as a useful synthetic method:
1)
MBH reaction has several advantages as a useful synthetic method:
1)
![Implications on Asymmetric Catalysis[edit] Nonetheless, the Aggarwal model shed light](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/397906/slide-23.jpg)
Implications on Asymmetric Catalysis[edit]
Nonetheless, the Aggarwal model shed light on the
Implications on Asymmetric Catalysis[edit]
Nonetheless, the Aggarwal model shed light on the


Sila-MBH reaction
Sila-MBH reaction is a MBH variant that couples α-silylated vinyl
Sila-MBH reaction
Sila-MBH reaction is a MBH variant that couples α-silylated vinyl
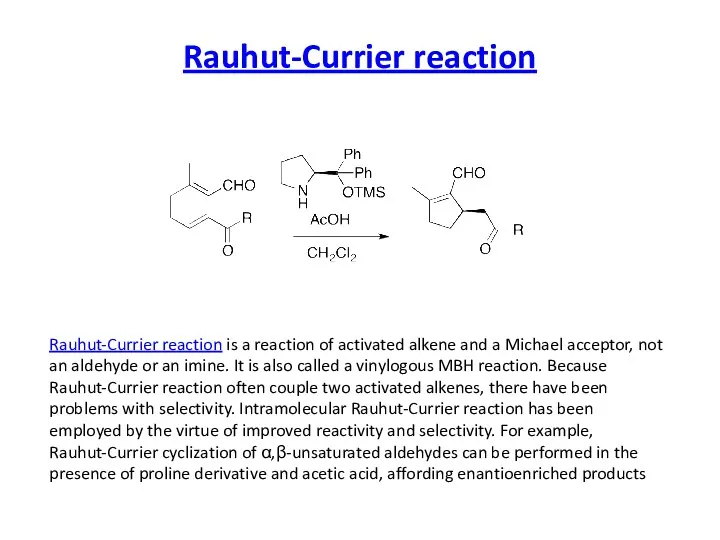
Rauhut-Currier reaction
Rauhut-Currier reaction is a reaction of activated alkene and a
Rauhut-Currier reaction
Rauhut-Currier reaction is a reaction of activated alkene and a

Tandem reaction / Multicomponent one-pot reaction
Multicomponent reaction strategy is attractive for
Tandem reaction / Multicomponent one-pot reaction
Multicomponent reaction strategy is attractive for
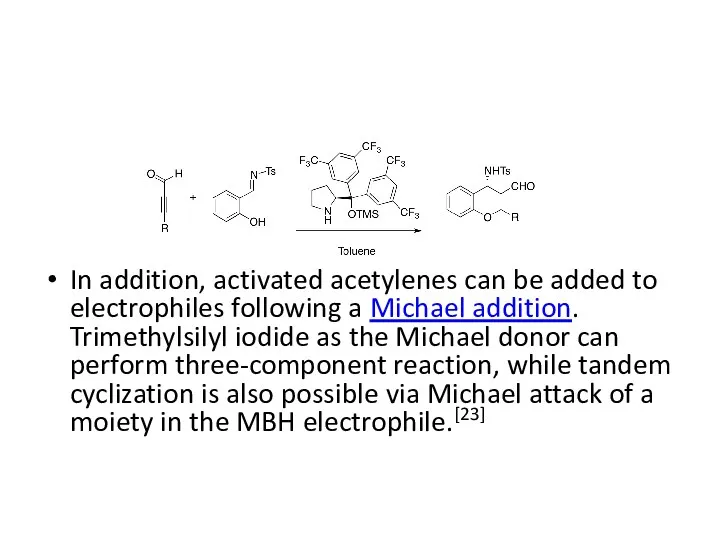
In addition, activated acetylenes can be added to electrophiles following a
In addition, activated acetylenes can be added to electrophiles following a
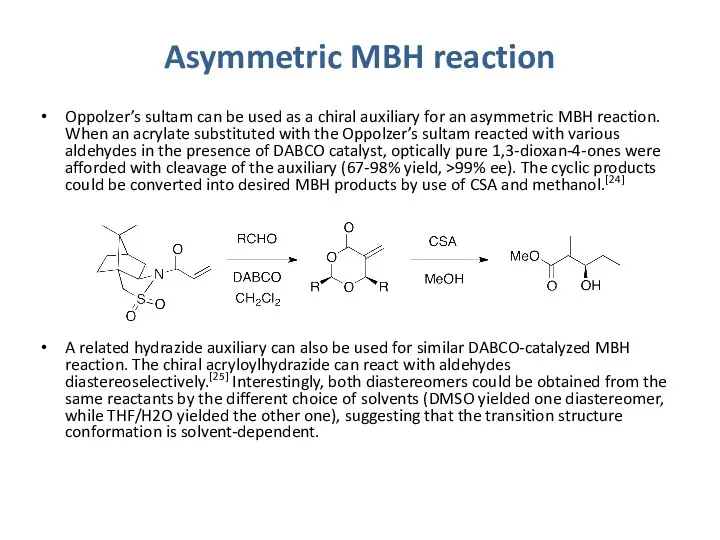
Asymmetric MBH reaction
Oppolzer’s sultam can be used as a chiral auxiliary
Asymmetric MBH reaction
Oppolzer’s sultam can be used as a chiral auxiliary
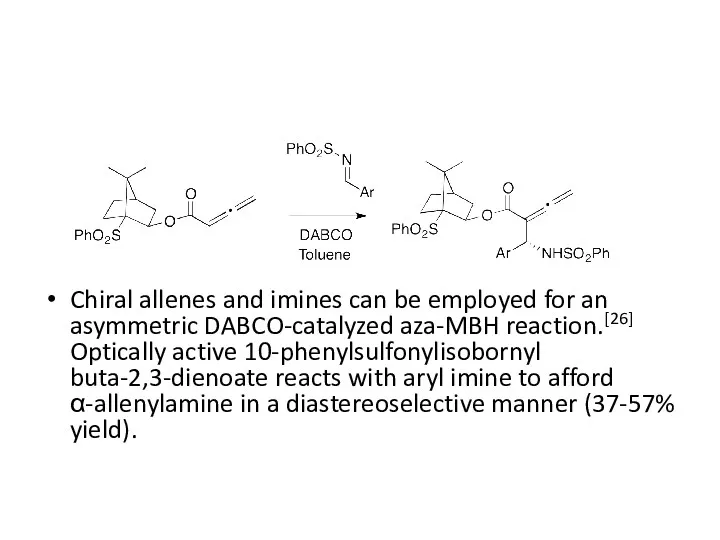
Chiral allenes and imines can be employed for an asymmetric DABCO-catalyzed
Chiral allenes and imines can be employed for an asymmetric DABCO-catalyzed

Chiral Lewis base catalyst
Chiral tertiary amine catalysts are employed for enantioselective
Chiral Lewis base catalyst
Chiral tertiary amine catalysts are employed for enantioselective

Simple diamine molecules can also be employed as MBH catalysts. Methyl
Simple diamine molecules can also be employed as MBH catalysts. Methyl

Chiral phosphine MBH catalysts
Chiral phosphine MBH catalysts often contain Bronsted acid
Chiral phosphine MBH catalysts
Chiral phosphine MBH catalysts often contain Bronsted acid
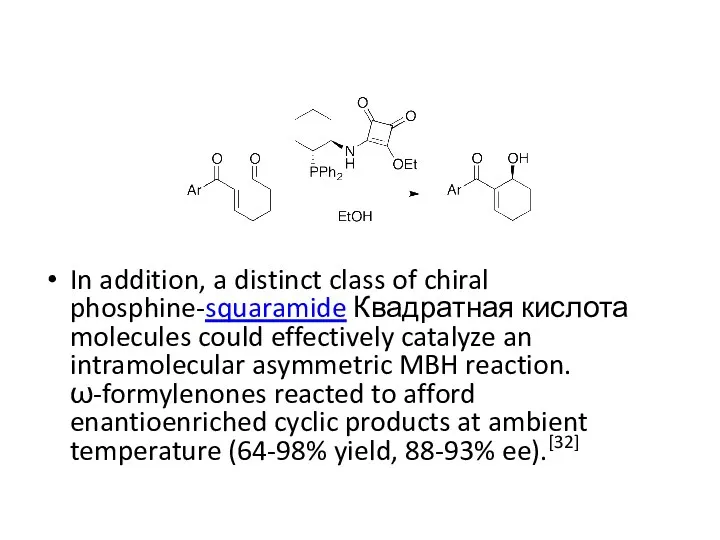
In addition, a distinct class of chiral phosphine-squaramide Квадратная кислота molecules
In addition, a distinct class of chiral phosphine-squaramide Квадратная кислота molecules
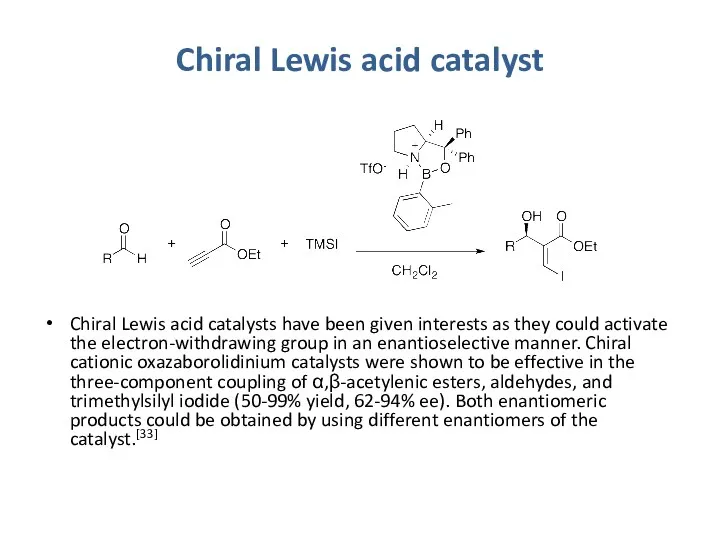
Chiral Lewis acid catalyst
Chiral Lewis acid catalysts have been given interests
Chiral Lewis acid catalyst
Chiral Lewis acid catalysts have been given interests
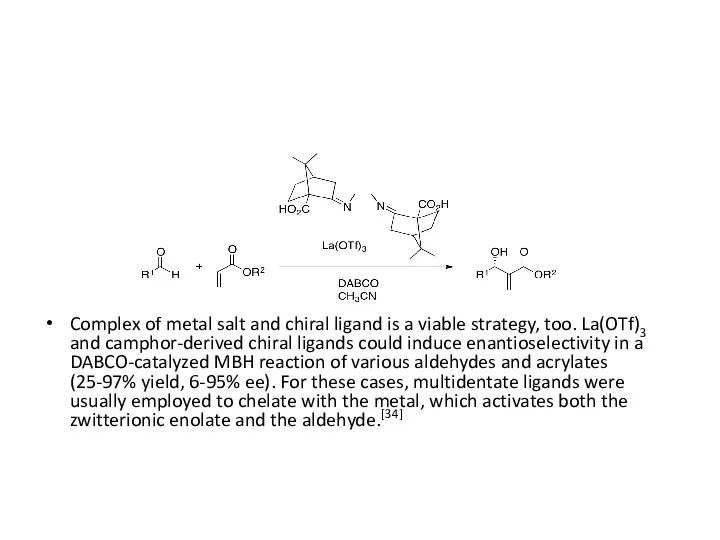
Complex of metal salt and chiral ligand is a viable strategy,
Complex of metal salt and chiral ligand is a viable strategy,
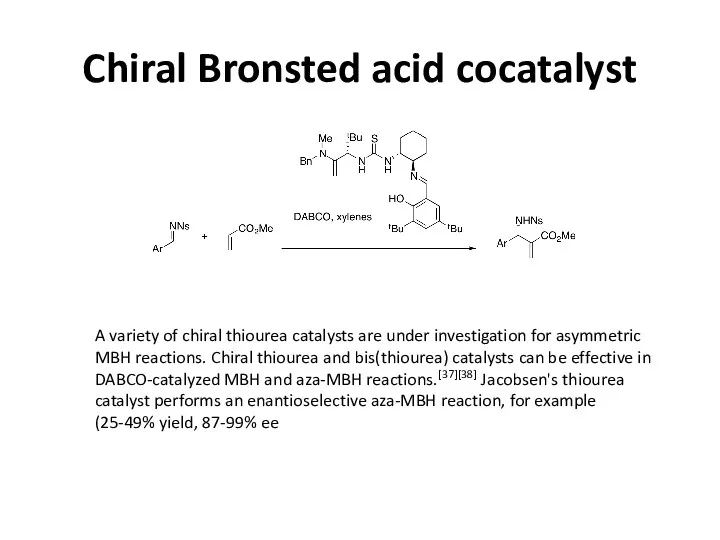
Chiral Bronsted acid cocatalyst
A variety of chiral thiourea catalysts are under
Chiral Bronsted acid cocatalyst
A variety of chiral thiourea catalysts are under

While simple thiourea requires a nucleophilic catalyst in conjunction, bifunctional catalysts
While simple thiourea requires a nucleophilic catalyst in conjunction, bifunctional catalysts
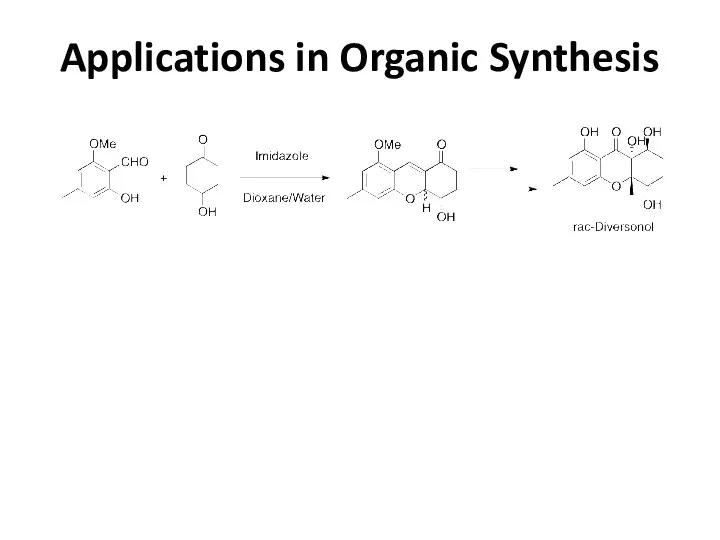
Applications in Organic Synthesis
Applications in Organic Synthesis
 Поняття про багатоатомні спирти на прикладі гліцеролу, його хімічні властивості
Поняття про багатоатомні спирти на прикладі гліцеролу, його хімічні властивості Анализ проб воды
Анализ проб воды Полиамиды. Классификация по методу получения
Полиамиды. Классификация по методу получения Кислород
Кислород Физико-химические и электро-физические свойства А3В5. Сравнительный анализ
Физико-химические и электро-физические свойства А3В5. Сравнительный анализ Молярный объем газов
Молярный объем газов fosfor_i_ego_soedineniya
fosfor_i_ego_soedineniya Қазақстанда химияны оқыту әдістемесінің даму тарихы
Қазақстанда химияны оқыту әдістемесінің даму тарихы Ферум та його сполуки
Ферум та його сполуки Элементтер-органогендер. Көміртек – органикалық қосылыстардың негізін құраушы. Тірі және өлі табиғат арасындағы шекара
Элементтер-органогендер. Көміртек – органикалық қосылыстардың негізін құраушы. Тірі және өлі табиғат арасындағы шекара Окислительно-восстановительные реакции
Окислительно-восстановительные реакции Маңызды мұнай өнімдері
Маңызды мұнай өнімдері Биополимеры
Биополимеры Энтальпия. Тепловой эффект химической реакции. 11 класс
Энтальпия. Тепловой эффект химической реакции. 11 класс Гидролиз солей
Гидролиз солей Лекция Атомное строение твердых тел. 1-01
Лекция Атомное строение твердых тел. 1-01 Стойкие органические загрязнители
Стойкие органические загрязнители Легированные стали
Легированные стали Химические и физические явления в жизни человека
Химические и физические явления в жизни человека Кристаллы. Выращивание кристалла
Кристаллы. Выращивание кристалла Химические волокна. Полиэфирные волокна. Лавсан
Химические волокна. Полиэфирные волокна. Лавсан Времена алхимиков
Времена алхимиков Альдегіди. Склад, будова молекул альдегідів. Альдегідна характеристична (функціональна) група
Альдегіди. Склад, будова молекул альдегідів. Альдегідна характеристична (функціональна) група Дистиляттағы цианидтер, алифаттық қатардағы галоген туындылары, хлороформ, хлоралгидрат, төртхлорлы көміртек
Дистиляттағы цианидтер, алифаттық қатардағы галоген туындылары, хлороформ, хлоралгидрат, төртхлорлы көміртек NaHSO4. Гидросульфат натрия
NaHSO4. Гидросульфат натрия Additives for Polymeric Materials
Additives for Polymeric Materials Карбоновые кислоты и их функциональные производные. Хроматографические методы исследования
Карбоновые кислоты и их функциональные производные. Хроматографические методы исследования Көмірсутектерді пиролиздеу арқылы қарапайым олефиндерді алу
Көмірсутектерді пиролиздеу арқылы қарапайым олефиндерді алу