Государственная служба Украины по лекарственным средствам. Установление критериев приемлемости. Аспекты валидации очистки презентация
- Государственная служба Украины по лекарственным средствам. Установление критериев приемлемости. Аспекты валидации очистки

Содержание
- 2. Введение Одним из ключевых моментов валидации очистки является расчет критериев чистоты оборудования (критериев приемлемости). Степень риска
- 3. Почему настолько важно правильно рассчитать критерии? уровень чистоты будет соответствовать требованиям методика контроля будет разработана на
- 4. Обзор требований Руководство 42-4.0-2013, Приложение 15, раздел Валидация очистки PI 006-2 RECOMMENDATION on VALIDATION MASTER PLAN,
- 5. Обзор требований …риск случайной перекрестной контаминации возникает … вследствие наличия остатков в оборудовании… Степень риска меняется
- 6. Обзор требований Наиболее опасной является контаминация препаратов, предназначенных для инъекций, а также препаратов, принимаемых в больших
- 7. Рекомендации PIC/S
- 8. Рекомендации PIC/S Валидация очистки 7.11 Установка допустимых пределов Обоснование выбранных пределов должно логически основываться на свойствах
- 9. Рекомендации PIC/S Могут применяться подходы для установки пределов: Валидация очистки для каждого продукта Группировка на семейства
- 10. Рекомендации PIC/S 7.11.3 Перенос остатков продукта должен отвечать определенным критериям, например, наиболее строгому из этих трех
- 11. Рекомендации PIC/S Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим методом в пробах, взятых
- 12. Рекомендации PIC/S Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим методом в пробах, взятых
- 13. Подходы к установке критериев На основе Терапевтической Дозы (ТД) и фактора безопасности (SF) a) не более
- 14. Критерий на основе доз Основан на допущении переноса определенной доли суточной дозы продукта в последующий продукт
- 15. Критерий на основе доз Многие фармацевтические компании используют стандартный фактор безопасности 1/1000 для всех расчетов предельных
- 16. Критерий на основе доз Это допущение не работает, когда перекрестное загрязнение переходит в лекарственную форму с
- 17. Порядок расчета критериев MACO мг/серию
- 18. Порядок расчета критериев SF – фактор безопасности По рекомендации PIC/S - не более 1/1000 или 0,1%
- 19. Критерии PDA TR №29 0,1%
- 20. Порядок расчета критериев
- 21. Порядок расчета критериев
- 22. Порядок расчета критериев При переходе А - B MACO = 2000 мг При переходе B -
- 23. Порядок расчета критериев МАСО ???=?? ???×(???/??×???) ???
- 24. Подходы к установке критериев b) не более 10 ррm любого продукта может появиться в следующем продукте
- 25. Порядок расчета критериев Устанавливается Максимально Допустимый Перенос (MACO) MACO = MBS [мг] х MAXCONC [мг] Кол-во
- 26. Порядок расчета критериев Пример 3: Очистка от продукта В. макс. суточная доза 250 мг, размер серии
- 27. Порядок расчета критериев На основе токсичности остатка (загрязнения) Основан на расчете т.н. числа NOEL (No Observed
- 28. Порядок расчета критериев Терапевтическая доза продукта может быть не известна. Поэтому используют расчет исходя из данных
- 29. Порядок расчета критериев
- 30. Порядок расчета критериев MACO мг/серию
- 31. Порядок расчета критериев Известны токсичность, объем серии и доза. МАСО можно рассчитать следующим образом. LD 50
- 32. Подходы к установке критериев Пероральное введение NOEL = LD 50 х 0,0005 х AAW = 419
- 33. Порядок расчета критериев Критерий, основанный на пределе аналитического обнаружения остатка Этот метод используется для таких специфических
- 34. Порядок расчета критериев
- 35. Распределение предела между отдельными частями оборудования Площадь всего задействованного оборудования принимается за 100%. Кол-во загрязнений на
- 36. Порядок расчета критериев M.P мг/м2, мг/дм2 мг/см2
- 37. Порядок расчета критериев Зависимость предела аналитического метода (М.СОNС) от предела загрязнения (МАСО) Установление максимальных пределов загрязнения
- 38. Порядок расчета критериев M.CONC мг/дм3 мг/л ррм
- 39. Интерпретация требований (пределы загрязнений) M.P мг/м2, мг/дм2 мг/см2 M.CONC мг/л (ррм) M.EQ мг/ед.обору- дования MACO мг/серию
- 40. Заключение Расчет пределов загрязнений на поверхности оборудования необходимо рассматривать только комплексно с учетом методов отбора проб.
- 42. Скачать презентацию

Введение
Одним из ключевых моментов валидации очистки является расчет критериев чистоты оборудования
Введение
Одним из ключевых моментов валидации очистки является расчет критериев чистоты оборудования

Почему настолько важно правильно рассчитать критерии?
уровень чистоты будет соответствовать требованиям
методика контроля
Почему настолько важно правильно рассчитать критерии?
уровень чистоты будет соответствовать требованиям
методика контроля

Обзор требований
Руководство 42-4.0-2013, Приложение 15, раздел Валидация очистки
PI 006-2 RECOMMENDATION on
Обзор требований
Руководство 42-4.0-2013, Приложение 15, раздел Валидация очистки
PI 006-2 RECOMMENDATION on

Обзор требований
…риск случайной перекрестной контаминации возникает … вследствие наличия остатков в
Обзор требований
…риск случайной перекрестной контаминации возникает … вследствие наличия остатков в

Обзор требований
Наиболее опасной является контаминация препаратов, предназначенных для инъекций, а
Обзор требований
Наиболее опасной является контаминация препаратов, предназначенных для инъекций, а

Рекомендации PIC/S
Рекомендации PIC/S

Рекомендации PIC/S
Валидация очистки
7.11 Установка допустимых пределов
Обоснование выбранных пределов должно логически основываться
Рекомендации PIC/S
Валидация очистки
7.11 Установка допустимых пределов
Обоснование выбранных пределов должно логически основываться

Рекомендации PIC/S
Могут применяться подходы для установки пределов:
Валидация очистки для каждого продукта
Группировка
Рекомендации PIC/S
Могут применяться подходы для установки пределов:
Валидация очистки для каждого продукта
Группировка

Рекомендации PIC/S
7.11.3 Перенос остатков продукта должен отвечать определенным критериям, например, наиболее
Рекомендации PIC/S
7.11.3 Перенос остатков продукта должен отвечать определенным критериям, например, наиболее

Рекомендации PIC/S
Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим
Рекомендации PIC/S
Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим

Рекомендации PIC/S
Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим
Рекомендации PIC/S
Определенные аллергены и сильнодействующие не должны обнаруживаться наилучшим возможным аналитическим

Подходы к установке критериев
На основе Терапевтической Дозы (ТД) и фактора безопасности
Подходы к установке критериев
На основе Терапевтической Дозы (ТД) и фактора безопасности

Критерий на основе доз
Основан на допущении переноса определенной доли суточной дозы
Критерий на основе доз
Основан на допущении переноса определенной доли суточной дозы

Критерий на основе доз
Многие фармацевтические компании используют стандартный фактор безопасности 1/1000
Критерий на основе доз
Многие фармацевтические компании используют стандартный фактор безопасности 1/1000

Критерий на основе доз
Это допущение не работает, когда перекрестное загрязнение переходит
Критерий на основе доз
Это допущение не работает, когда перекрестное загрязнение переходит

Порядок расчета критериев
MACO
мг/серию
Порядок расчета критериев
MACO
мг/серию

Порядок расчета критериев
SF – фактор безопасности
По рекомендации PIC/S - не
Порядок расчета критериев
SF – фактор безопасности
По рекомендации PIC/S - не

Критерии PDA TR №29
0,1%
Критерии PDA TR №29
0,1%

Порядок расчета критериев
Порядок расчета критериев

Порядок расчета критериев
Порядок расчета критериев

Порядок расчета критериев
При переходе А - B MACO = 2000 мг
При
Порядок расчета критериев
При переходе А - B MACO = 2000 мг
При

Порядок расчета критериев
МАСО ???=?? ???×(???/??×???) ???
Порядок расчета критериев
МАСО ???=?? ???×(???/??×???) ???

Подходы к установке критериев
b) не более 10 ррm любого продукта может
Подходы к установке критериев
b) не более 10 ррm любого продукта может

Порядок расчета критериев
Устанавливается Максимально Допустимый Перенос (MACO)
MACO = MBS [мг] х
Порядок расчета критериев
Устанавливается Максимально Допустимый Перенос (MACO)
MACO = MBS [мг] х

Порядок расчета критериев
Пример 3: Очистка от продукта В.
макс. суточная доза
Порядок расчета критериев
Пример 3: Очистка от продукта В.
макс. суточная доза

Порядок расчета критериев
На основе токсичности остатка (загрязнения)
Основан на расчете т.н. числа
Порядок расчета критериев
На основе токсичности остатка (загрязнения)
Основан на расчете т.н. числа

Порядок расчета критериев
Терапевтическая доза продукта может быть не известна. Поэтому используют
Порядок расчета критериев
Терапевтическая доза продукта может быть не известна. Поэтому используют

Порядок расчета критериев
Порядок расчета критериев

Порядок расчета критериев
MACO
мг/серию
Порядок расчета критериев
MACO
мг/серию

Порядок расчета критериев
Известны токсичность, объем серии и доза. МАСО можно рассчитать
Порядок расчета критериев
Известны токсичность, объем серии и доза. МАСО можно рассчитать

Подходы к установке критериев
Пероральное введение
NOEL = LD 50 х 0,0005
Подходы к установке критериев
Пероральное введение
NOEL = LD 50 х 0,0005

Порядок расчета критериев
Критерий, основанный на пределе аналитического обнаружения остатка
Этот метод
Порядок расчета критериев
Критерий, основанный на пределе аналитического обнаружения остатка
Этот метод

Порядок расчета критериев
Порядок расчета критериев

Распределение предела между
отдельными частями оборудования
Площадь всего задействованного оборудования принимается за
Распределение предела между
отдельными частями оборудования
Площадь всего задействованного оборудования принимается за

Порядок расчета критериев
M.P
мг/м2,
мг/дм2
мг/см2
Порядок расчета критериев
M.P
мг/м2,
мг/дм2
мг/см2

Порядок расчета критериев
Зависимость предела аналитического метода (М.СОNС) от предела загрязнения (МАСО)
Порядок расчета критериев
Зависимость предела аналитического метода (М.СОNС) от предела загрязнения (МАСО)

Порядок расчета критериев
M.CONC
мг/дм3
мг/л
ррм
Порядок расчета критериев
M.CONC
мг/дм3
мг/л
ррм
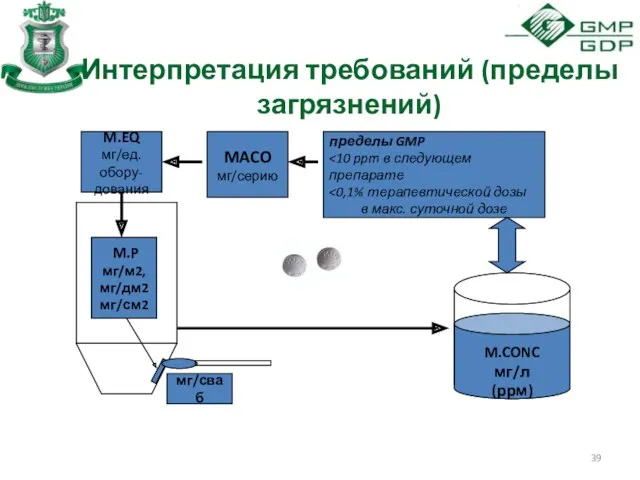
Интерпретация требований (пределы загрязнений)
M.P
мг/м2,
мг/дм2
мг/см2
M.CONC
мг/л
(ррм)
M.EQ
мг/ед.обору-
дования
MACO
мг/серию
пределы GMP
<10 ppm в следующем
Интерпретация требований (пределы загрязнений)
M.P
мг/м2,
мг/дм2
мг/см2
M.CONC
мг/л
(ррм)
M.EQ
мг/ед.обору-
дования
MACO
мг/серию
пределы GMP
<10 ppm в следующем

Заключение
Расчет пределов загрязнений на поверхности оборудования необходимо рассматривать только комплексно с
Заключение
Расчет пределов загрязнений на поверхности оборудования необходимо рассматривать только комплексно с
 Мешел ауруы
Мешел ауруы Внебольничные пневмонии и грипп
Внебольничные пневмонии и грипп Түйе етінің биологиялық құндылығы
Түйе етінің биологиялық құндылығы Секреты жизни
Секреты жизни Ювенильді ревматоидты артрит
Ювенильді ревматоидты артрит Клеточные повреждения. Клеточные повреждения, устойчивое нарушение клеточного гомеостазиса. Классификация клеточных повреждений
Клеточные повреждения. Клеточные повреждения, устойчивое нарушение клеточного гомеостазиса. Классификация клеточных повреждений Климактерический период
Климактерический период Производные фторхинолонов (ломефлоксацин, офлоксацин) и хиназолина (празозин). Требования к качеству, методы анализа
Производные фторхинолонов (ломефлоксацин, офлоксацин) и хиназолина (празозин). Требования к качеству, методы анализа Коммуникативтік дағдылар. Сырқатқа деген көзқарастың түрлері
Коммуникативтік дағдылар. Сырқатқа деген көзқарастың түрлері Качество пищевых продуктов. Стандартизация и сертификация
Качество пищевых продуктов. Стандартизация и сертификация Вегетарианство
Вегетарианство Обследование лиц с заиканием
Обследование лиц с заиканием Инфузионная терапия
Инфузионная терапия Мамандандырылған медициналық жәрдемді ұйымдастыру
Мамандандырылған медициналық жәрдемді ұйымдастыру Регулирование биологических лекарственных средств и их аналогов на примере эритропоэтинов
Регулирование биологических лекарственных средств и их аналогов на примере эритропоэтинов Лекарственные растения в домашней аптечке
Лекарственные растения в домашней аптечке Қоршаған орта мен халықтың денсаулығы
Қоршаған орта мен халықтың денсаулығы Реабилитациядағы нормативті-құқықты актілер
Реабилитациядағы нормативті-құқықты актілер Геморрагический инсульт
Геморрагический инсульт Сепсис. Характеристика заболевания
Сепсис. Характеристика заболевания Основы патопсихологии
Основы патопсихологии Врачебные ошибки и ятрогении в деятельности медицинских работников
Врачебные ошибки и ятрогении в деятельности медицинских работников Вогнепальні та невогнепальні пошкодження щелеп
Вогнепальні та невогнепальні пошкодження щелеп Основные клинические синдромы в пульмонологии
Основные клинические синдромы в пульмонологии Артериальная гипертензия у детей и подростков
Артериальная гипертензия у детей и подростков PBSerum. Инновации в космецевтике
PBSerum. Инновации в космецевтике Вещества, влияющие на сосудистый тонус
Вещества, влияющие на сосудистый тонус Гострі кишкові інфекції у дітей (1-ша частина)
Гострі кишкові інфекції у дітей (1-ша частина)