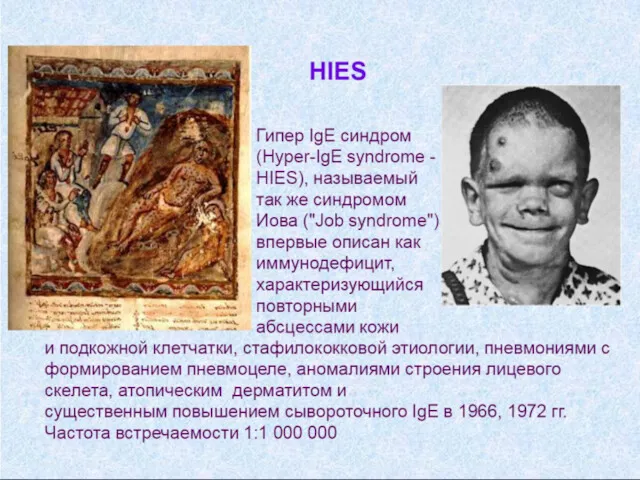
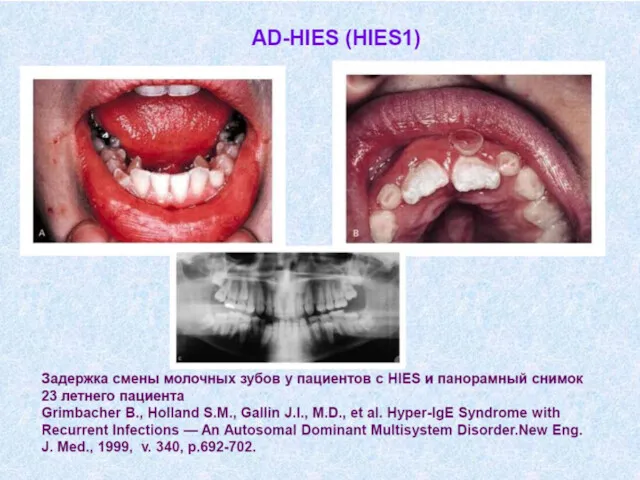
- Иммунодефицитные состояния у детей

Содержание
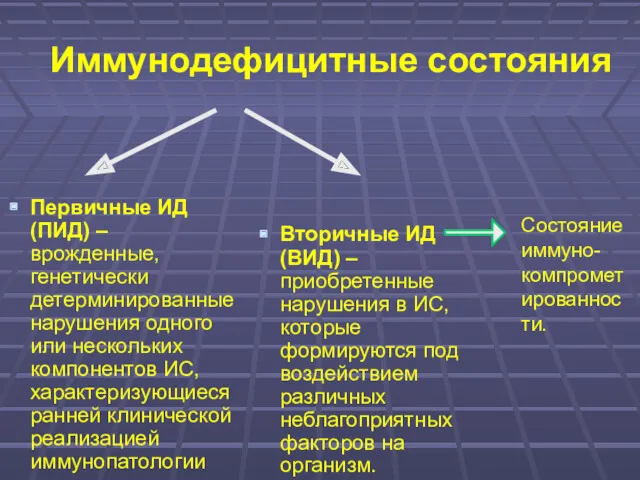
- 2. Иммунодефицитные состояния Первичные ИД (ПИД) – врожденные, генетически детерминированные нарушения одного или нескольких компонентов ИС, характеризующиеся
- 3. Сопоставление понятий «иммунокомпрометированный ребенок» и вторичное ИДС
- 4. В последние годы сформировалось отчетливое представление о том, что ПИД – более частое состояние, чем это
- 5. ПИД – проблема педиатрическая: Манифестация чаще происходит в детском возрасте. Первые формы известных ПИД были описаны
- 6. Актуальность темы определяется следующими положениями: В педиатрической практике существует гиподиагностика истинных ИДС. Так, описаны случаи постановки
- 7. Причины гиподиагностики: Методы молекулярно-генетического анализа для диагностики ИДС малодоступны в повседневной клинической практике. На сегодняшнем этапе
- 8. Повсеместное, в т.ч. коммерческое использование нестандартных, неадекватных, устаревших методов исследования. На сегодняшний день самым точным методом
- 9. Первичные ИДС. (ПИДС) Относительно редкие заболевания с преобладающим аутосомно-рецессивным типом наследования. Многие классические формы ПИДС сцеплены
- 10. Рабочая классификация ПИДС (2006г.) Объединяет ~120 синдромов ПИДС. В зависимости от уровня нарушения и локализации дефекта
- 11. 1. Комбинированный ИД – генетический дефект, препятствующий нормальной дифференцировке стволовой клетки или предшественников Т- лимфоцитов. Основные
- 49. 5. Дефекты системы комплемента. Дефект любого из 9ти компонентов отсутствие снижение количества Дефициты С5 – С9
- 50. Ассоциированные с иммунодефицитом синдромы: Ифекционный Желудочно-кишечный неинфекционного генеза Лимфопролиферативный ∕ онкологический Аллергический Аутоиммунный
- 51. Особенности инфекционного синдрома: -собирательное понятие, включает в себя инфекционно-воспалительные заболевания различной этиологии (бактериальной, вирусной грибковой, микоплазменной,
- 52. - раннее, быстрое присоединение условно-патогенной микрофлоры; - ведущая микст инфекция в формировании воспалительного процесса; - необычные
- 53. Лимфопролиферативный синдром Склонность к опухолевому росту у пациентов с ПИДС (лимфоретикулярные злокачественные опухоли) Аллергический синдром Дефект
- 54. Аутоимунный синдром: Агаммаглобулинемия, дефекты системы комплемента ▼ Поражение соединительной Ткани Дерматомиозит Склеродермия Аутоимунный гепатит ЮРА Аутоимунный
- 55. Первичные иммунодефицитные состояния - 10 настораживающих признаков: 1. Частые заболевания отитом (не менее 6-8 раз в
- 56. Первичные иммунодефицитные состояния - 10 настораживающих признаков: 6. Потребность во внутривенных антибиотиках для купирования инфекции+ повтор
- 58. Скачать презентацию
Иммунодефицитные состояния
Первичные ИД (ПИД) – врожденные, генетически детерминированные нарушения одного или
Иммунодефицитные состояния
Первичные ИД (ПИД) – врожденные, генетически детерминированные нарушения одного или

Сопоставление понятий «иммунокомпрометированный ребенок» и вторичное ИДС
Сопоставление понятий «иммунокомпрометированный ребенок» и вторичное ИДС

В последние годы сформировалось отчетливое представление о том, что ПИД –
В последние годы сформировалось отчетливое представление о том, что ПИД –

ПИД – проблема педиатрическая:
Манифестация чаще происходит в детском возрасте.
Первые формы известных
ПИД – проблема педиатрическая:
Манифестация чаще происходит в детском возрасте.
Первые формы известных

Актуальность темы определяется следующими положениями:
В педиатрической практике существует гиподиагностика истинных ИДС.
Актуальность темы определяется следующими положениями:
В педиатрической практике существует гиподиагностика истинных ИДС.

Причины гиподиагностики:
Методы молекулярно-генетического анализа для диагностики ИДС малодоступны в повседневной клинической
Причины гиподиагностики:
Методы молекулярно-генетического анализа для диагностики ИДС малодоступны в повседневной клинической

Повсеместное, в т.ч. коммерческое использование нестандартных, неадекватных, устаревших методов исследования. На
Повсеместное, в т.ч. коммерческое использование нестандартных, неадекватных, устаревших методов исследования. На

Первичные ИДС. (ПИДС)
Относительно редкие заболевания с преобладающим аутосомно-рецессивным типом наследования.
Многие
Первичные ИДС. (ПИДС)
Относительно редкие заболевания с преобладающим аутосомно-рецессивным типом наследования.
Многие

Рабочая классификация ПИДС (2006г.)
Объединяет ~120 синдромов ПИДС.
В зависимости от уровня нарушения
Рабочая классификация ПИДС (2006г.)
Объединяет ~120 синдромов ПИДС.
В зависимости от уровня нарушения

1. Комбинированный ИД – генетический дефект, препятствующий нормальной дифференцировке стволовой клетки
1. Комбинированный ИД – генетический дефект, препятствующий нормальной дифференцировке стволовой клетки
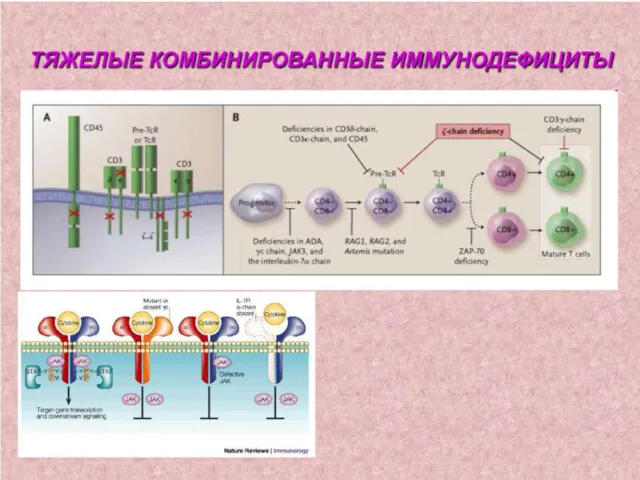
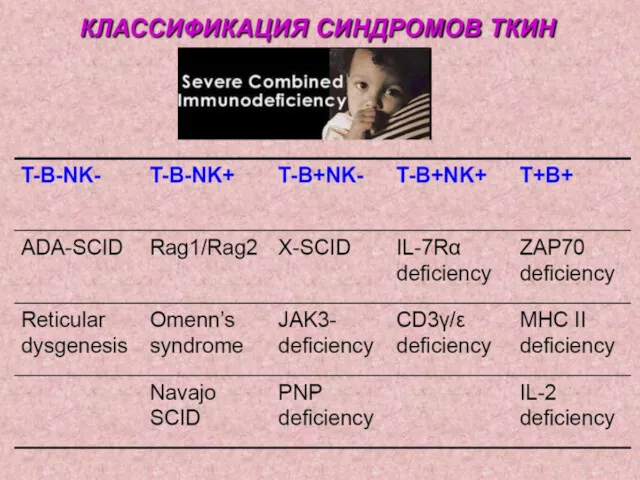









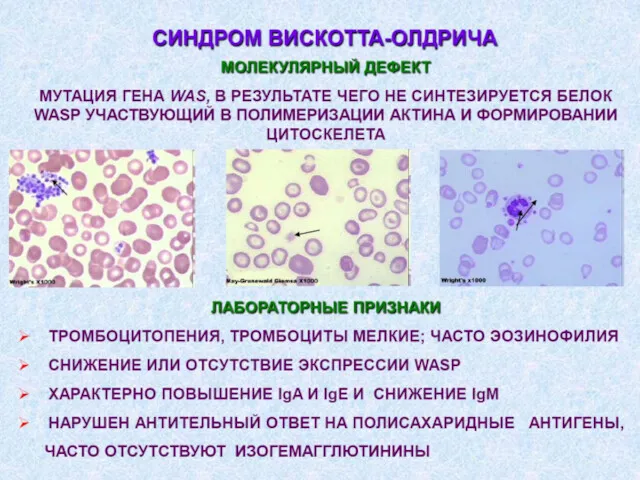


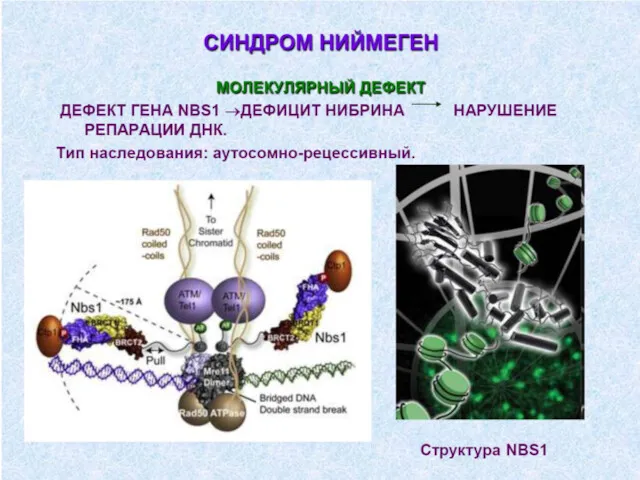










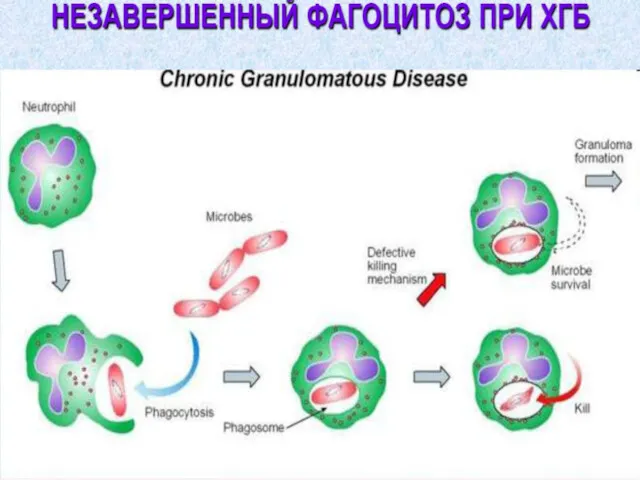








5. Дефекты системы комплемента.
Дефект любого из 9ти компонентов
отсутствие снижение количества
Дефициты
5. Дефекты системы комплемента.
Дефект любого из 9ти компонентов
отсутствие снижение количества
Дефициты

Ассоциированные с иммунодефицитом синдромы:
Ифекционный
Желудочно-кишечный неинфекционного генеза
Лимфопролиферативный ∕ онкологический
Аллергический
Аутоиммунный
Ассоциированные с иммунодефицитом синдромы:
Ифекционный
Желудочно-кишечный неинфекционного генеза
Лимфопролиферативный ∕ онкологический
Аллергический
Аутоиммунный

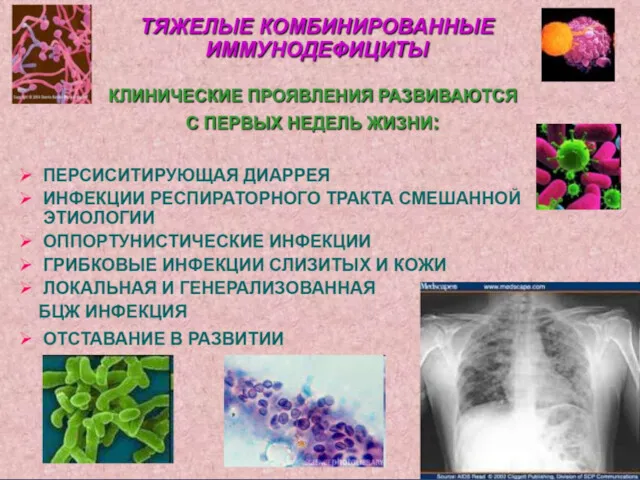
Особенности инфекционного синдрома:
-собирательное понятие, включает в себя инфекционно-воспалительные заболевания различной этиологии
Особенности инфекционного синдрома:
-собирательное понятие, включает в себя инфекционно-воспалительные заболевания различной этиологии

- раннее, быстрое присоединение условно-патогенной микрофлоры;
- ведущая микст инфекция в формировании
- раннее, быстрое присоединение условно-патогенной микрофлоры;
- ведущая микст инфекция в формировании

Лимфопролиферативный синдром
Склонность к опухолевому росту у пациентов с ПИДС
(лимфоретикулярные злокачественные опухоли)
Аллергический
Лимфопролиферативный синдром
Склонность к опухолевому росту у пациентов с ПИДС
(лимфоретикулярные злокачественные опухоли)
Аллергический

Аутоимунный синдром:
Агаммаглобулинемия, дефекты системы
комплемента
▼
Поражение соединительной
Ткани
Дерматомиозит Склеродермия
Аутоимунный гепатит ЮРА Аутоимунный
Аутоимунный синдром:
Агаммаглобулинемия, дефекты системы
комплемента
▼
Поражение соединительной
Ткани
Дерматомиозит Склеродермия
Аутоимунный гепатит ЮРА Аутоимунный

Первичные иммунодефицитные состояния
- 10 настораживающих признаков:
1. Частые заболевания отитом (не менее
Первичные иммунодефицитные состояния
- 10 настораживающих признаков:
1. Частые заболевания отитом (не менее

Первичные иммунодефицитные состояния
- 10 настораживающих признаков:
6. Потребность во внутривенных антибиотиках для
Первичные иммунодефицитные состояния
- 10 настораживающих признаков:
6. Потребность во внутривенных антибиотиках для
 Министр здравоохранения ДНР - Долгошапко Ольга Николаевна
Министр здравоохранения ДНР - Долгошапко Ольга Николаевна Метаболічні перетворення білків і амінокислот
Метаболічні перетворення білків і амінокислот Drugs affecting efferent innervation cholinomimetics, anticholinesterase drugs
Drugs affecting efferent innervation cholinomimetics, anticholinesterase drugs Лекарственные средства
Лекарственные средства Clinical anatomy of abdominal cavity
Clinical anatomy of abdominal cavity Эхинококкоз
Эхинококкоз Лучевая диагностика заболеваний органов грудной полости
Лучевая диагностика заболеваний органов грудной полости Радиацияның табиғаты және оның биологиялық әрекеті
Радиацияның табиғаты және оның биологиялық әрекеті Антидепрессанты
Антидепрессанты Физиология эндокринной системы
Физиология эндокринной системы Гемостаз. Физиология системы крови. Лекция 3
Гемостаз. Физиология системы крови. Лекция 3 Химико-токсикологический анализ наркотических веществ
Химико-токсикологический анализ наркотических веществ Показатели для оценки физического развития ребенка в различные возрастные периоды. Паспорт здоровья ребенка
Показатели для оценки физического развития ребенка в различные возрастные периоды. Паспорт здоровья ребенка Общая фармакология
Общая фармакология Лечебные и профилактические зубные пасты
Лечебные и профилактические зубные пасты Судебно-медицинская экспертиза вещественных доказательств
Судебно-медицинская экспертиза вещественных доказательств Молекулярно-генетическая диагностика колоректального рака
Молекулярно-генетическая диагностика колоректального рака Программы профилактики в стоматологии
Программы профилактики в стоматологии Острый тонзиллит
Острый тонзиллит Клиникалық жағдай
Клиникалық жағдай Основы психопатологии. Психиатрия
Основы психопатологии. Психиатрия Беременность и новорожденный. Интереснейшие факты про беременность
Беременность и новорожденный. Интереснейшие факты про беременность Способы лечения варикозной болезни нижних конечностей
Способы лечения варикозной болезни нижних конечностей Физиология высшей нервной деятельности
Физиология высшей нервной деятельности Оказание акушерской помощи
Оказание акушерской помощи Саркоидоз легких
Саркоидоз легких Хирургическая инфекция. Острая гнойная инфекция кожи, клетчаточных пространств
Хирургическая инфекция. Острая гнойная инфекция кожи, клетчаточных пространств Инсульт. Первая медицинская помощь
Инсульт. Первая медицинская помощь