- Врожденная миотония Томсена. Миотоническая дистрофия

Содержание
- 2. Содержание: Определение заболевания Классификация заболевания Механизм развития Фенотип больного Клинические проявления Диагностика и лечение Список использованной
- 3. Определение заболевания Миотония - (myotoniae; греч. mys, myos мышца + tonos напряжение) – нервно-мышечное заболевание, характеризующееся
- 4. Классификация заболевания: OMIM: 160800 МКБ 11: -G 71.1 Первичные поражения мышц клиническая: первые признаки заболевания возникают
- 5. Механизм развития: Наследуется по аутосомно-доминантному типу Мутация в гене CLCN1 Нарушение Расслабления мышц Снижение проницаемости плазмолеммы
- 6. https://kiberis.ru/?p=34217 Миотонический спазм круговой мышцы глаз Фенотип больного Ноги ребенка при болезни Томсена https://medicalplanet.su/neurology/bolezn_ tomsena_u_detei.html
- 7. Клинические проявления: В начальной стадии: проявляется изменением голоса, особенно при плаче, ребенок начинает задыхаться, а после
- 8. Диагностика: Диагноз строится на основании генеалогического анализа (аутосомно-доминантный тип наследования), особенностей клинической картины (атлетический тип телосложения,
- 9. Определение заболевания Миотоническая дистрофия —Наследственное мультисистемное заболевание из группы нервно-мышечных заболеваний, при котором нарушается нормальное функционирование
- 10. Классификация заболевания: OMIM: тип 1 (160900) тип 2 (602668). МКБ 11 G71.1. Миотонические расстройства клиническая Заболевание
- 11. Механизм развития: Наследуется по аутосомно-доминантному типу Мутация в гене DMPK Поражение скелетных мышц, нарушение проводимости сердца,
- 12. http://medicfoto.ru/post.php?id=358 Миотония Россолимо- Куршмана-Штейнерта-Баттена Фенотип больного
- 13. Клинические проявления: В начальной стадии: ДМ представлены мышечными (миопатия, миотония, миалгия) и внемышечными симпто- мами, среди
- 14. Диагностика: Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа.
- 16. Скачать презентацию

Содержание:
Определение заболевания
Классификация заболевания
Механизм развития
Фенотип больного
Клинические проявления
Диагностика и лечение
Список использованной литературы
Содержание:
Определение заболевания
Классификация заболевания
Механизм развития
Фенотип больного
Клинические проявления
Диагностика и лечение
Список использованной литературы

Определение заболевания
Миотония - (myotoniae; греч. mys, myos мышца + tonos напряжение)
Определение заболевания
Миотония - (myotoniae; греч. mys, myos мышца + tonos напряжение)

Классификация заболевания:
OMIM: 160800
МКБ 11:
-G 71.1 Первичные поражения мышц
клиническая:
первые признаки заболевания возникают
Классификация заболевания:
OMIM: 160800
МКБ 11:
-G 71.1 Первичные поражения мышц
клиническая:
первые признаки заболевания возникают
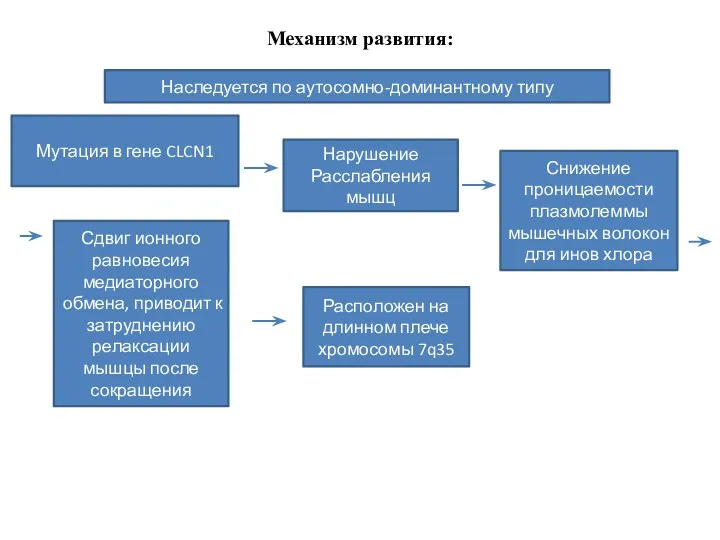
Механизм развития:
Наследуется по аутосомно-доминантному типу
Мутация в гене CLCN1
Нарушение
Расслабления мышц
Снижение проницаемости
Механизм развития:
Наследуется по аутосомно-доминантному типу
Мутация в гене CLCN1
Нарушение
Расслабления мышц
Снижение проницаемости
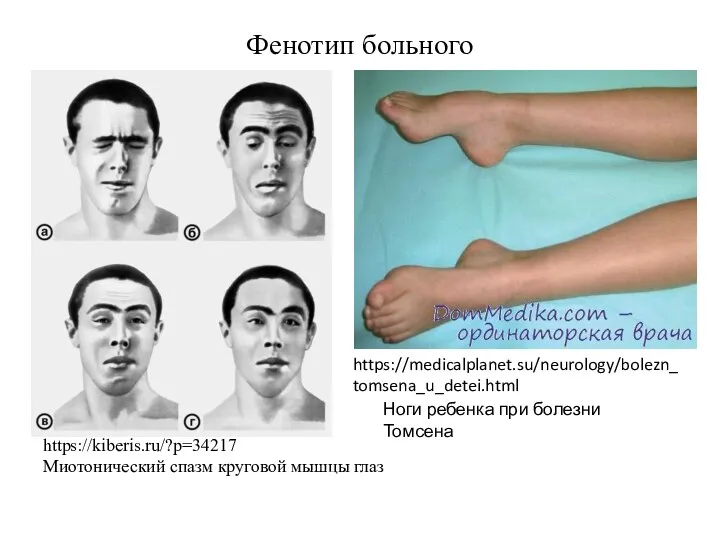
https://kiberis.ru/?p=34217
Миотонический спазм круговой мышцы глаз
Фенотип больного
Ноги ребенка при болезни Томсена
https://medicalplanet.su/neurology/bolezn_
tomsena_u_detei.html
https://kiberis.ru/?p=34217
Миотонический спазм круговой мышцы глаз
Фенотип больного
Ноги ребенка при болезни Томсена
https://medicalplanet.su/neurology/bolezn_
tomsena_u_detei.html

Клинические проявления:
В начальной стадии: проявляется изменением голоса, особенно при плаче, ребенок
Клинические проявления:
В начальной стадии: проявляется изменением голоса, особенно при плаче, ребенок

Диагностика:
Диагноз строится на основании генеалогического анализа (аутосомно-доминантный тип наследования), особенностей клинической
Диагностика: Диагноз строится на основании генеалогического анализа (аутосомно-доминантный тип наследования), особенностей клинической

Определение заболевания
Миотоническая дистрофия —Наследственное мультисистемное заболевание из группы нервно-мышечных заболеваний, при
Определение заболевания
Миотоническая дистрофия —Наследственное мультисистемное заболевание из группы нервно-мышечных заболеваний, при

Классификация заболевания:
OMIM: тип 1 (160900)
тип 2 (602668).
МКБ 11
G71.1. Миотонические расстройства
клиническая
Классификация заболевания:
OMIM: тип 1 (160900)
тип 2 (602668).
МКБ 11
G71.1. Миотонические расстройства
клиническая
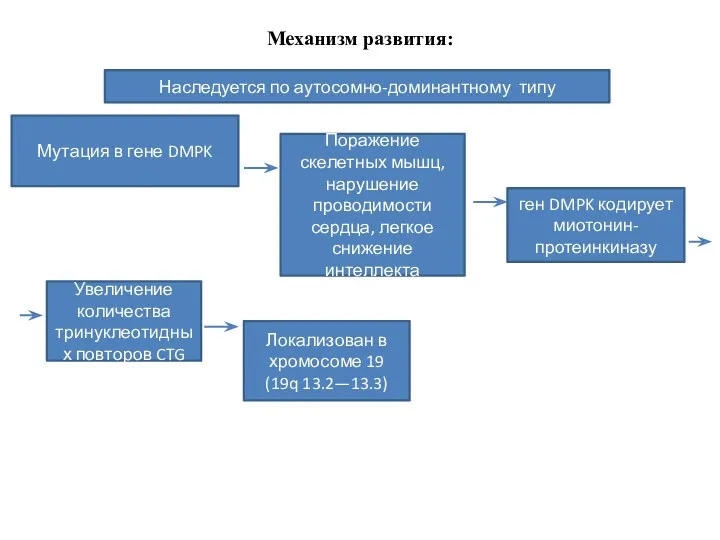
Механизм развития:
Наследуется по аутосомно-доминантному типу
Мутация в гене DMPK
Поражение скелетных мышц,
Механизм развития:
Наследуется по аутосомно-доминантному типу
Мутация в гене DMPK
Поражение скелетных мышц,
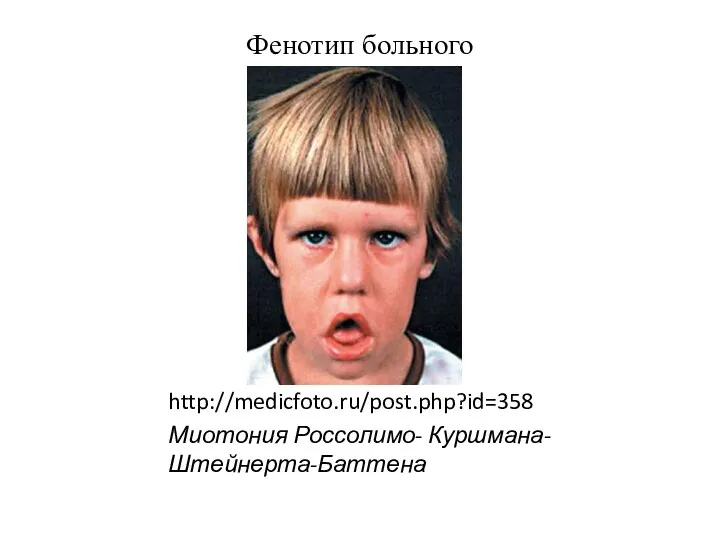
http://medicfoto.ru/post.php?id=358
Миотония Россолимо- Куршмана-Штейнерта-Баттена
Фенотип больного
http://medicfoto.ru/post.php?id=358
Миотония Россолимо- Куршмана-Штейнерта-Баттена
Фенотип больного

Клинические проявления:
В начальной стадии: ДМ представлены мышечными (миопатия, миотония, миалгия) и
Клинические проявления:
В начальной стадии: ДМ представлены мышечными (миопатия, миотония, миалгия) и

Диагностика:
Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе
Диагностика: Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе
 Заболевания органов пищеварения
Заболевания органов пищеварения Организация кабинета медицинского массажа. (Тема 1.1)
Организация кабинета медицинского массажа. (Тема 1.1) Топографическая анатомия и оперативная хирургия тонкой и толстой кишки
Топографическая анатомия и оперативная хирургия тонкой и толстой кишки Проблемы боли в детской стоматологии. Принципы анестезиологии в детской стоматологии
Проблемы боли в детской стоматологии. Принципы анестезиологии в детской стоматологии Первичный туберкулез у детей: патогенез, особенности, первичной туберкулезной инфекции
Первичный туберкулез у детей: патогенез, особенности, первичной туберкулезной инфекции Тұқым қуалаушылықтың патологиядағы рөлі
Тұқым қуалаушылықтың патологиядағы рөлі TRƯỜNG ĐAI HỌC BÁCH KHOA TP.HCM KHOA KHOA HỌC ỨNG DỤNG MÔN CƠ SỞ KỸ THUẬT Y SINH
TRƯỜNG ĐAI HỌC BÁCH KHOA TP.HCM KHOA KHOA HỌC ỨNG DỤNG MÔN CƠ SỞ KỸ THUẬT Y SINH Лейкоцитозы, лейкопении, лейкозы. Принципы классификации, механизмы развития клинических и гематологических признаков
Лейкоцитозы, лейкопении, лейкозы. Принципы классификации, механизмы развития клинических и гематологических признаков Белок. Правильное питание как необходимое условие здоровья и долголетия
Белок. Правильное питание как необходимое условие здоровья и долголетия Геморрагиялық диатездер
Геморрагиялық диатездер Рак яичника (клиника, диагностика, лечение)
Рак яичника (клиника, диагностика, лечение) Синдром Гийена-Барре
Синдром Гийена-Барре Бешенство (гидрофобия)
Бешенство (гидрофобия) mol
mol Анаэробные инфекции. Клостридии. Клостридиозы
Анаэробные инфекции. Клостридии. Клостридиозы Токсичность и опасность химических веществ. Токсикометрия
Токсичность и опасность химических веществ. Токсикометрия Хеликобактер. Классификация. Заболевания, вызываемые ими. Микробиология
Хеликобактер. Классификация. Заболевания, вызываемые ими. Микробиология Особенности реализации плана системного ухода за больными взрослыми и детьми терапевтического профиля
Особенности реализации плана системного ухода за больными взрослыми и детьми терапевтического профиля 20230925_osanka
20230925_osanka Постхолецистэктомический синдром
Постхолецистэктомический синдром Ветряная оспа. Паротит
Ветряная оспа. Паротит Хронические облитерирующие заболевания артерий нижних конечностей
Хронические облитерирующие заболевания артерий нижних конечностей Женский таз и тазовое дно
Женский таз и тазовое дно Рациональная антибиотикотерапия в акушерско-гинекологической практике
Рациональная антибиотикотерапия в акушерско-гинекологической практике Учение об инфекции патогенность и вирулентность микробов
Учение об инфекции патогенность и вирулентность микробов Неотложные состояния в практике медицинской сестры
Неотложные состояния в практике медицинской сестры Мытье и антисептика рук персонала ЛПУ
Мытье и антисептика рук персонала ЛПУ Мультикиназные ингибиторы - 2
Мультикиназные ингибиторы - 2