- Группа компаний Р-ФАРМ. Инновационные технологии здоровья

Содержание
- 2. О компании Основана в 2001 году Вертикально-интегрированная компания, являющаяся одним из лидеров рынка Продуктовый портфель состоит
- 3. Ключевые инвестиции в проекты за пределами России Турция, 2013 Открыт филиал Индия, 2010 Запущен партнерский R&D
- 4. Целевой профиль препарата FDA guidance: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm080593.pdf
- 5. Разработка препаратов – поэтапный процесс оценки эффективности и безопасности как у животных, так и у человека
- 6. Основные пути создания лекарственных средств. 1.Модификация структур известных лекарственных средств. 2. Копирование известных физиологически активных веществ.
- 7. Создание лекарственного препарата — длительный процесс В создании новых ЛС участвуют представители многих профессий: химики, биологи,
- 8. Полусинтез многие сложные природные вещества на основе биологически активных полупродуктов естественного происхождения, например полусинтетические пенициллины: ампициллин,
- 9. Биосинтез Моноклональные антитела –вырабатываются иммунными клетками. Моноклональные антитела могут быть выработаны против почти любого природного антигена
- 10. Генная инженерия Целенаправленное изменение генетических программ клеток с целью видоизменения или создания принципиально новых форм микроорганизмов,
- 11. Примеры генной инженерии 1. Инсулин . 2. Микробный продуцент соматотропного гормона роста человека— соматотропин, дает возможность
- 12. Виды ЛС Оригинальное лекарственное средство - лекарственное средство (ЛС), содержащее впервые полученную фармацевтическую субстанцию или новую
- 13. Биоподобное ЛС «биоаналоги» или «биоподобные препараты» («biosimilars»): это лекарственные средства, произведенные путем биотехнологических процессов с применением:
- 14. Молекулярная сложность биопрепаратов / PRESENTATION
- 15. Отличия биологических препаратов от препаратов химического синтеза 1. Большой молекулярный вес 2. Сложность пространственной структуры белка:
- 16. Основополагающая разница между химическими и биотехнологическими препаратами Химические препараты Простые химические компоненты Однотипная прогнозируемая структура, которую
- 17. Причины различий оригинальных и воспроизведенных ЛС Фармацевтическая технология производства препарата Вспомогательные вещества (неактивные ингредиенты, наполнители, консерванты
- 18. Сравнение стоимости оригинальных и воспроизведенных препаратов 80% стоимости оригинального ЛС – стоимость исследований эффективности и безопасности
- 19. Параметры оценки эквивалентности дженериков
- 20. Программа клинических исследований Исследования фармакокинетики, доклинической токсичности 15
- 21. Клиническое исследование изучение клинических, фармакологических, фармакодинамических свойств исследуемого препарата у человека, включая процессы всасывания, распределения, изменения
- 22. История возникновения законодательной базы клинических исследований Датой возникновения регуляторной базы можно считать 1937 г. Компания M.
- 23. 1 фаза клинических исследований – это «ворота» между научным исследованием и клиникой. 1.Риск от препарата и
- 24. ФАЗЫ КЛИНИЧЕСКОГО ИСПЫТАНИЯ I фаза клинического исследования относится первоначальное введение человеку нового экспериментального лекарственного средства. Главная
- 25. II фаза клинических исследований оценка эффективности воздействия лекарственного средства в связи с конкретным показанием или показаниями
- 26. III фаза клинического исследования Их главная задача - получить дополнительные сведения по эффективности и безопасности различных
- 28. Скачать презентацию

О компании
Основана в 2001 году
Вертикально-интегрированная компания, являющаяся одним из лидеров
О компании
Основана в 2001 году
Вертикально-интегрированная компания, являющаяся одним из лидеров
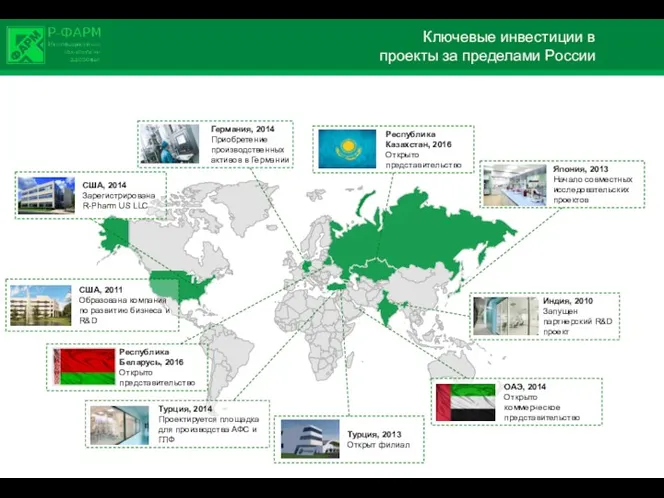
Ключевые инвестиции в
проекты за пределами России
Турция, 2013 Открыт филиал
Индия,
Ключевые инвестиции в
проекты за пределами России
Турция, 2013 Открыт филиал
Индия,

Целевой профиль препарата
FDA guidance:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm080593.pdf
Целевой профиль препарата
FDA guidance:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm080593.pdf
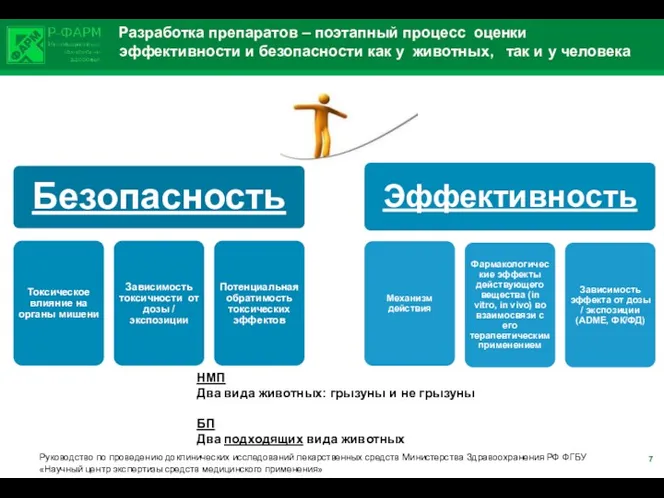
Разработка препаратов – поэтапный процесс оценки эффективности и безопасности как
Разработка препаратов – поэтапный процесс оценки эффективности и безопасности как

Основные пути
создания лекарственных средств.
1.Модификация структур известных лекарственных средств.
2. Копирование известных
Основные пути
создания лекарственных средств.
1.Модификация структур известных лекарственных средств.
2. Копирование известных

Создание лекарственного препарата — длительный процесс
В создании новых ЛС участвуют
Создание лекарственного препарата — длительный процесс
В создании новых ЛС участвуют

Полусинтез
многие сложные природные вещества на основе биологически активных полупродуктов естественного происхождения,
Полусинтез
многие сложные природные вещества на основе биологически активных полупродуктов естественного происхождения,

Биосинтез
Моноклональные антитела –вырабатываются иммунными клетками. Моноклональные антитела могут быть выработаны против
Биосинтез
Моноклональные антитела –вырабатываются иммунными клетками. Моноклональные антитела могут быть выработаны против

Генная инженерия
Целенаправленное изменение генетических программ клеток с целью видоизменения или создания
Генная инженерия
Целенаправленное изменение генетических программ клеток с целью видоизменения или создания

Примеры генной инженерии
1. Инсулин .
2. Микробный продуцент соматотропного гормона
Примеры генной инженерии
1. Инсулин .
2. Микробный продуцент соматотропного гормона

Виды ЛС
Оригинальное лекарственное средство - лекарственное средство (ЛС), содержащее впервые полученную
Виды ЛС
Оригинальное лекарственное средство - лекарственное средство (ЛС), содержащее впервые полученную

Биоподобное ЛС
«биоаналоги» или «биоподобные препараты» («biosimilars»):
это лекарственные средства, произведенные путем биотехнологических
Биоподобное ЛС
«биоаналоги» или «биоподобные препараты» («biosimilars»):
это лекарственные средства, произведенные путем биотехнологических
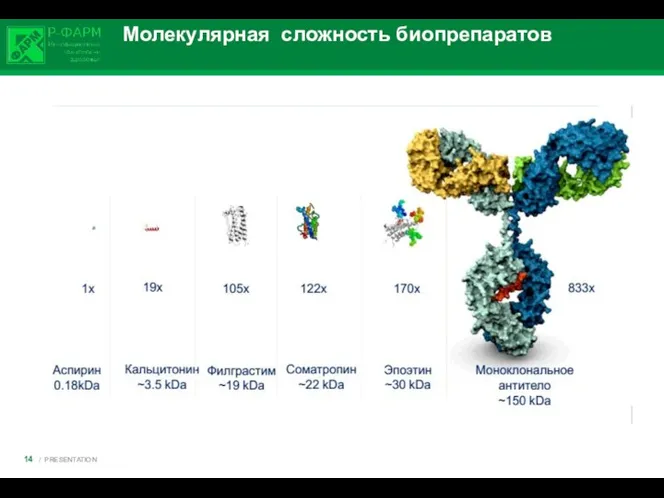
Молекулярная сложность биопрепаратов
/ PRESENTATION
Молекулярная сложность биопрепаратов
/ PRESENTATION
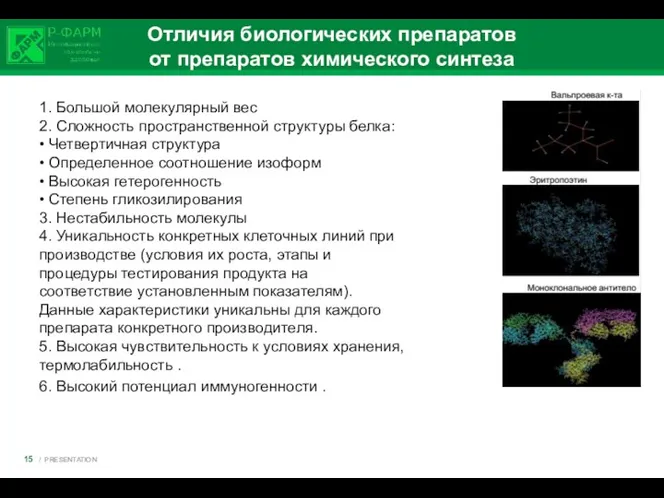
Отличия биологических препаратов
от препаратов химического синтеза
1. Большой молекулярный вес
2. Сложность
Отличия биологических препаратов
от препаратов химического синтеза
1. Большой молекулярный вес 2. Сложность

Основополагающая разница между химическими и биотехнологическими препаратами
Химические препараты
Простые химические компоненты
Однотипная прогнозируемая
Основополагающая разница между химическими и биотехнологическими препаратами
Химические препараты
Простые химические компоненты
Однотипная прогнозируемая

Причины различий оригинальных
и воспроизведенных ЛС
Фармацевтическая технология производства препарата
Вспомогательные вещества (неактивные
Причины различий оригинальных
и воспроизведенных ЛС
Фармацевтическая технология производства препарата
Вспомогательные вещества (неактивные

Сравнение стоимости оригинальных
и воспроизведенных препаратов
80% стоимости оригинального ЛС –
Сравнение стоимости оригинальных
и воспроизведенных препаратов
80% стоимости оригинального ЛС –

Параметры оценки
эквивалентности дженериков
Параметры оценки
эквивалентности дженериков
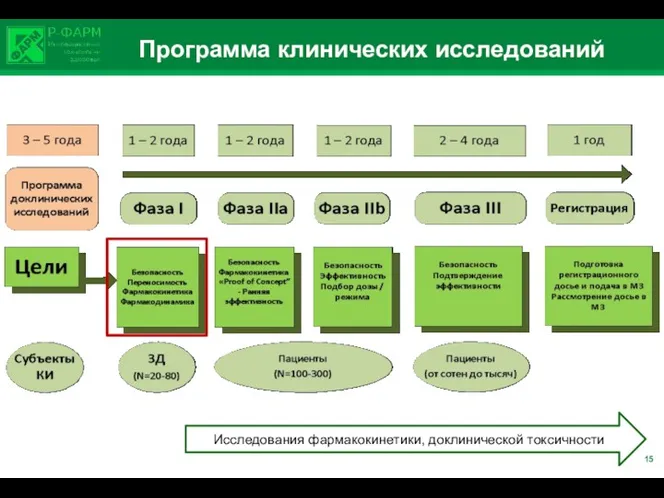
Программа клинических исследований
Исследования фармакокинетики, доклинической токсичности
15
Программа клинических исследований
Исследования фармакокинетики, доклинической токсичности
15

Клиническое исследование
изучение клинических, фармакологических, фармакодинамических свойств исследуемого препарата у человека, включая
Клиническое исследование
изучение клинических, фармакологических, фармакодинамических свойств исследуемого препарата у человека, включая

История возникновения законодательной
базы клинических исследований
Датой возникновения регуляторной базы можно считать
История возникновения законодательной
базы клинических исследований
Датой возникновения регуляторной базы можно считать

1 фаза клинических исследований – это «ворота» между научным исследованием
1 фаза клинических исследований – это «ворота» между научным исследованием

ФАЗЫ КЛИНИЧЕСКОГО ИСПЫТАНИЯ
I фаза клинического исследования относится первоначальное введение человеку нового
ФАЗЫ КЛИНИЧЕСКОГО ИСПЫТАНИЯ
I фаза клинического исследования относится первоначальное введение человеку нового

II фаза клинических исследований
оценка эффективности воздействия лекарственного средства в связи
II фаза клинических исследований
оценка эффективности воздействия лекарственного средства в связи

III фаза клинического исследования
Их главная задача - получить дополнительные сведения
III фаза клинического исследования
Их главная задача - получить дополнительные сведения
 Методы и инструментарий государственного регулирования, экономики
Методы и инструментарий государственного регулирования, экономики Тема 6. Рынок. Конкуренция. Монополия
Тема 6. Рынок. Конкуренция. Монополия Introduction to macroeconomics (Lecture 1)
Introduction to macroeconomics (Lecture 1) Основные производственные фонды
Основные производственные фонды Соціально - економічне становище в Україні в 1953-1964 рр
Соціально - економічне становище в Україні в 1953-1964 рр Основные отрасли промышлености Кировской области
Основные отрасли промышлености Кировской области Узбекистан. Международная торговля
Узбекистан. Международная торговля Понятие, содержание и субъекты экономической деятельности и экономических отношений
Понятие, содержание и субъекты экономической деятельности и экономических отношений Расчет основных технико-экономических показателей работ, связанных с электроснабжением добычного участка
Расчет основных технико-экономических показателей работ, связанных с электроснабжением добычного участка Прибыль и рентабельность производства
Прибыль и рентабельность производства Постсоветское пространство. Укрепление влияния России и его кризис
Постсоветское пространство. Укрепление влияния России и его кризис Екологічна та енергетична проблеми у світі. Способи їх вирішення
Екологічна та енергетична проблеми у світі. Способи їх вирішення ТЭК управляет миром
ТЭК управляет миром Международные индексы и рейтинги, структура и содержание
Международные индексы и рейтинги, структура и содержание Экономиканың тұжырымдамасы. Макроэкономикалық көрсеткіштер: ұлттық табыс (жiө, жұө), жұмыспен қамту, экономика
Экономиканың тұжырымдамасы. Макроэкономикалық көрсеткіштер: ұлттық табыс (жiө, жұө), жұмыспен қамту, экономика Основы бизнес-планирования
Основы бизнес-планирования Теория прибавочной стоимости. Карл Маркс
Теория прибавочной стоимости. Карл Маркс Теория фирмы
Теория фирмы Экономические системы. Подготовка к ЕГЭ. Обществознание
Экономические системы. Подготовка к ЕГЭ. Обществознание Қазақстан Республикасы мен Түркия мемлекетінің экономикалық жағдайы
Қазақстан Республикасы мен Түркия мемлекетінің экономикалық жағдайы ВВП: определение, методы расчета. Система национальных счетов
ВВП: определение, методы расчета. Система национальных счетов Уровень цены
Уровень цены Бизнес-модель логистического распределительного центра в Оренбургской области
Бизнес-модель логистического распределительного центра в Оренбургской области Демографічна проблема
Демографічна проблема Труд - основа жизни
Труд - основа жизни Модель системы налогообложения. (Лекция 2)
Модель системы налогообложения. (Лекция 2) Анализ подходов к формированию финансовой политики предприятий лесоперерабатывающей отрасли в условиях эконом. нестабильности
Анализ подходов к формированию финансовой политики предприятий лесоперерабатывающей отрасли в условиях эконом. нестабильности Продовольственная безопасность в республике Беларусь в условиях экономических санкций
Продовольственная безопасность в республике Беларусь в условиях экономических санкций