- Начала химической термодинамики

Содержание
- 3. Фаза - отдельная часть системы, отделенная от других ее частей хотя бы одной поверхностью раздела кристалл
- 4. термодинамические параметры Интенсивные П.: величина не зависит от количества вещества (Т, P, СМ) Экстенсивные П.: величина
- 5. МГУ
- 6. При проведении процесса считаем, что каждое небольшое изменение параметров сопровождается установлением т/д равновесия, так что процесс
- 7. В химических реакциях часто выделяется или поглощается теплота В физических процессах: плавление требует подвода теплоты извне,
- 8. На что израсходовалась подведенная теплота Q в указанном примере? работа расширения пара: A = P·ΔV работа
- 9. Абсолютные значения U и H определить невозможно. Но: нас интересует энергетический эффект процесса, т.е. изменение состояния
- 10. Термохимия - раздел химической т/д, изучающий теплоты химических реакций термохимические уравнения - агрегатное состояние (!) -
- 11. При проведении реакции в изобарных условиях тепловой эффект реакции определяется изменением энтальпии (Qp = ΔH), то
- 12. Тепловые эффекты можно рассчитывать! Закон Гесса (II закон термохимии – также частный случай закона сохранения материи
- 13. ΔН(1) – энтальпия образования CO (г) ΔН(2) – энтальпия образования CO2 (г) Следствие из закона Гесса
- 14. Стандартная энтальпия образования простых веществ принята равной нулю Хлор – газообразный, Cl2 Сера – твердое вещество,
- 15. Схема (а) и энтальпийная диаграмма (б) для изменения энтальпии реакции газообразного NH3 с раствором HCl ΔНx
- 16. Энергия связи -энергия, которую необходимо затратить на разрыв связи А-В ΔН0A-B -энтальпия образования связи А-В можно
- 17. Атомарная теплота образования (Δa.f.Н0ABn) -энтальпия образования данного вещества ABn из атомов ABn = A + nB
- 18. ΔНреш.NaCl = ΔНобр - ΔНсубл - ΔНсв - ΔНион – ΔНе Цикл Борна-Габера: U0 Энергия кристаллической
- 19. Энтальпийная диаграмма цикла Борна-Габера для NaCl
- 20. Энергия некоторых кристаллических решеток Ахметов, с.185 (U0298)
- 21. Изменением энтальпии можно охарактеризовать многие процессы Энтальпия (теплота) сгорания (combustion) - важная характеристика топлив и пищи
- 22. 3. Процессы в растворах Термохимические расчеты в растворах проводят не по теплотам образования молекул, а по
- 23. Для H+(aq), исходя из ΔfН0(298) = 0, найдено ΔhН0(298) = -1075 кДж/моль Энтальпии гидратации отдельных ионов
- 24. Растворение ионного соединения состоит из двух стадий – разрушения кристаллической решетки на свободные ионы и их
- 25. Выше для расчета энтальпии реакции использовали стандартные энтальпии образования продуктов и исходных веществ. Энтальпию реакции можно
- 26. Кроме того, энтальпию сгорания можно использовать для расчета стандартной энтальпии образования вещества (!) СxНyOz(г) + (x+y/4-z/2)
- 27. по энергиям связи (DA-B) (например, когда неизвестна теплота образования одного из реагентов) Результат приблизительный, поскольку используются
- 28. Температурная зависимость энтальпии До сих пор рассматривались процессы, где температура исходных веществ и продуктов была одинакова
- 29. При этом сама теплоемкость сложным образом зависит от температуры ! Зависимость Ср вещества от Т. Вдали
- 30. Пример зависимости от Т теплоемкости исх. в-в (1) и продуктов (2) Пример зависимости от Т энтальпии
- 31. Если изучаемый температурный интервал невелик, и в нем не происходит фазовый переход, то можно записать упрощенную
- 32. Направления процессов в физико-химических системах До XIX века полагали, что вещества реагируют, если имеют сродство друг
- 33. Мы уже можем рассчитать ΔrH, не прибегая к эксперименту. А можно ли установить принципиальную возможность и
- 34. Sº(298) растет при переходе тв – ж – г в аморфном состоянии выше, чем в кристаллическом
- 35. При нагревании вещества его энтропия возрастает, причем при температурах фазовых переходов происходят скачки Прирост энтропии при
- 36. По данным о стандартной энтропии вещества можно рассчитать изменение энтропии химического процесса j - продукты i
- 37. Приложение функций состояния к установлению направления процесса Изменение энтальпии отражает стремление системы к взаимодействию (объединению частиц
- 38. Формулировка второго начала термодинамики для неизолированных систем: Невозможно осуществить перенос тепла от более холодного тела к
- 39. Возрастание энтропии в системе – «энтропийный фактор». Количественно оценивают в виде произведения TΔS (Дж/моль) Изменение энергии
- 40. В чем состоит физический смысл энтропии? Больцман (1896 г.) предложил определить это понятие с использованием статистической
- 41. G - энергия Гиббса (свободная энергия) - характеристика устойчивости системы при постоянном давлении Для учета совместного
- 42. ΔH = ΔG + TΔS Изменение энтальпии в процессе состоит из двух частей ΔH ΔG -
- 43. ΔG = (ΔН – TΔS) Для самопроизвольного протекания процесса при любых температурах необходимо сочетание При достаточно
- 44. 1 Для практических целей используют значения энергии Гиббса реакций для стандартных условий (ΔG0298) Для простых веществ,
- 45. Вещества, для которых ΔG0298 0, вещество термодинамически нестабильно. Внимание! Из ΔG0298 = 0 не следует, что
- 46. Третий путь определения стандартного изменения энергии Гиббса реакции основан на комбинации значений ΔrG0298 хорошо изученных реакций
- 47. Фримантл, с. 254 Диаграммы Эллингема позволяют наглядно определять, какой оксид будет восстанавливаться в каждой конкретной реакции
- 48. Третьяков, с. 31 Вероятность самопроизвольного протекания химических реакций При высоких температурах основную роль играет энтропийный фактор
- 49. Итак, многие химические процессы, начиная протекать в одном направлении, затем идут в обоих (вз-е продуктов), т.е
- 50. В обратимом процессе через некоторое время устанавливается состояние равновесия При постоянных внешних условиях (p,V,T,состав) – равновесное
- 51. Кажущееся равновесие - заторможенное (метастабильное состояние) К.р. сходно с Истинным р. по неизменности состояния во времени
- 52. Тогда для гомогенной реакции справедливо: aA + bB + … ⮀ dD + eE + …
- 53. В гетерогенных реакциях концентрации твердых фаз в уравнение константы равновесия не входят: CaCO3 (к) ⮀ CaO
- 54. Изменение концентраций (или парциальных давлений) влияют не на величину константы равновесия, а на степень превращения. Это
- 55. Влияние давления на равновесие - в системах, где есть изменение объема газообразных веществ Эффективность действия фактора
- 56. Влияние концентрации на равновесие В соответствии с принципом Ле-Шателье введение в равновесную систему дополнительных количеств какого-либо
- 57. Термодинамика дает важное соотношение (изотерма реакции): Изменения ΔG в реальных условиях химических реакций из которого для
- 58. Зависимость константы равновесия от температуры Преобразуем уравнение в и получим Карапетьянц с. 199
- 59. Объединяя и получаем и Найденную из эксперимента зависимость константы равновесия от температуры используют для расчета термодинамических
- 60. Скорость и механизм химических реакций Как мы выяснили, реакции с ΔG > 0 самопроизвольно не идут.
- 61. Реакция гомогенная (в объеме фазы, напр., в растворе) гетерогенная (на поверхности раздела фаз, напр., газ-твердое тело)
- 62. Пример: Третьяков, с.103 Для реакции 2Н2О2 = 2Н2О + О2 Изменение концентрации Н2О2 во времени. tgα
- 63. Скорость химической реакции зависит от многих факторов: природы реагирующих веществ концентрации реагирующих веществ температуры наличия катализатора
- 64. m и n равны стехиометрическим коэффициентам только в том случае, если уравнение реакции соответствует элементарной стадии,
- 65. Для реакции разложения Н2О2: v = k[Н2О2]1 Кинетические уравнения для реакций с различным порядком 2Н2О2 =
- 66. На практике порядок реакции по реагенту определяют из графика в логарифмических координатах в соответствии с преобразованием
- 67. Физический смысл величины k: константа скорости реакции численно равна скорости реакции при единичных концентрациях реагентов v
- 68. Период полупревращения вещества - время, за которое прореагирует половина его количества Интегрированием дифференциальной формы кинетического уравнения
- 69. Итак: Только элементарные реакции идут так, как они записаны, сложные реакции (большинство) – это набор нескольких
- 70. «Кинетический» вывод константы равновесия Для обратимой одностадийной реакции А + В ⮀ АВ vпр = k[А][B]
- 71. Зависимость скорости реакции от температуры Третьяков, с.102 Поверхность потенциальной энергии системы H2 + Br Профиль потенциальной
- 72. Зависимость скорости реакции от температуры В подавляющем большинстве случаев скорость реакции с повышением температуры увеличивается. Пример:
- 73. «Температура ускоряет реакции, потому что молекулы чаще соударяются» неверный ответ! при ΔТ = 10º скорость движения
- 74. Физический смысл предэкспоненциального множителя А: A = Z·P Z – число, пропорциональное количеству соударений (зависит от
- 75. О влиянии температуры на скорость химической реакции с точки зрения молекулярно-кинетической теории Распределение частиц по энергиям
- 76. Катализ изменение скорости химической реакции в результате действия катализатора - вещества, изменяющего скорость определенной химической реакции,
- 77. без катализатора: с катализатором: SO2 + ½ O2 ⮀ SO3 Eакт = Еа1 NO + ½
- 78. ингибирование Автокаталитические процессы MO + 2 HF ⮀ MF2 + H2O появление воды резко ускоряет прямую
- 79. Гомогенный катализ в газовой фазе – рассмотрен выше на примере реакции SO2 → SO3 (Кт NO)
- 80. Гетерогенный катализ Процессы идут на границе раздела фаз, поэтому для объяснения привлекают теорию адсорбции Адсорбция –
- 81. Рассматривают пять стадий: 1. Диффузия
- 82. 2. Адсорбция
- 83. 3. Реакция
- 84. 4. Десорбция
- 85. 5. Диффузия
- 86. Активность катализатора зависит от присутствия посторонних веществ: промоторы – не обладают каталитической активностью, но увеличивают активность
- 87. Скорость реакции в гетерогенных системах Для реакций в гетерогенных системах очень важны процессы переноса Три основных
- 88. Кроме обратимых реакций известны и иные сложные процессы, состоящие из двух или более простых реакций: параллельные
- 89. если v1 то v = f(v1) лимитирующая стадия H2O2 2 OH* OH* + HI Пример: I
- 90. сопряженные реакции (химическая индукция) А+В M N А+C и когда одна из реакций индуцируется другой Пример:
- 91. Цепные реакции Связанная система сложных реакций, протекающих последовательно, параллельно и сопряженно с участием свободных радикалов, называется
- 93. Скачать презентацию
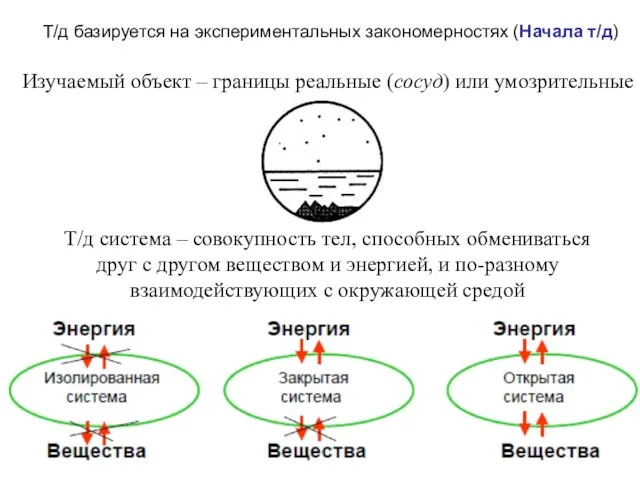
Фаза
- отдельная часть системы, отделенная от других ее частей хотя бы
Фаза
- отдельная часть системы, отделенная от других ее частей хотя бы
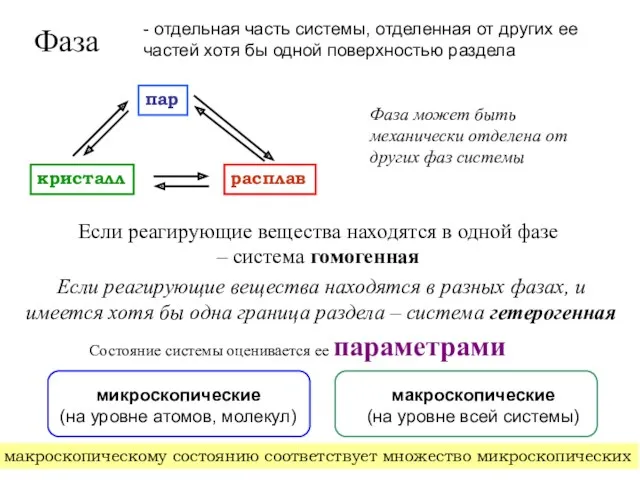
термодинамические параметры
Интенсивные П.: величина не зависит от количества вещества
(Т, P,
термодинамические параметры
Интенсивные П.: величина не зависит от количества вещества
(Т, P,
МГУ
МГУ

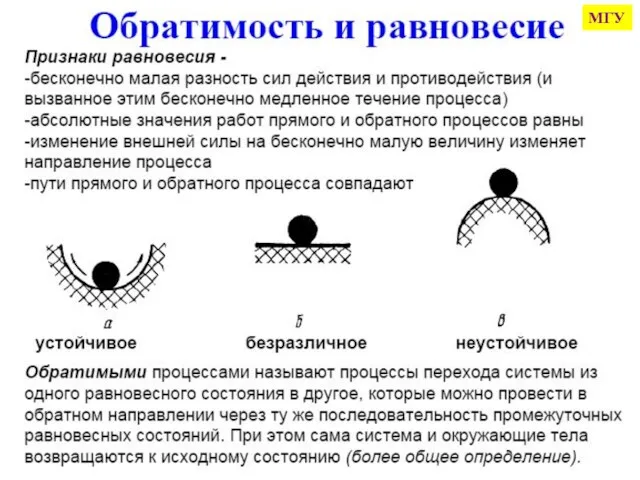
При проведении процесса считаем, что каждое небольшое изменение параметров сопровождается установлением
При проведении процесса считаем, что каждое небольшое изменение параметров сопровождается установлением

В химических реакциях часто выделяется или поглощается теплота
В физических процессах: плавление
В химических реакциях часто выделяется или поглощается теплота
В физических процессах: плавление
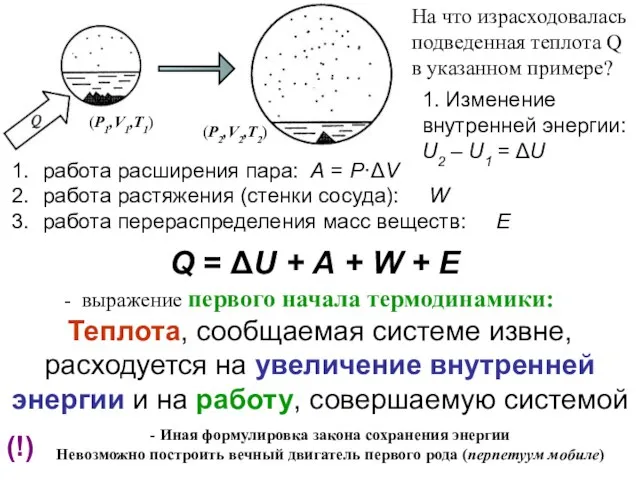
На что израсходовалась подведенная теплота Q в указанном примере?
работа расширения
На что израсходовалась подведенная теплота Q в указанном примере?
работа расширения
Абсолютные значения U и H определить невозможно. Но: нас интересует энергетический
Абсолютные значения U и H определить невозможно. Но: нас интересует энергетический
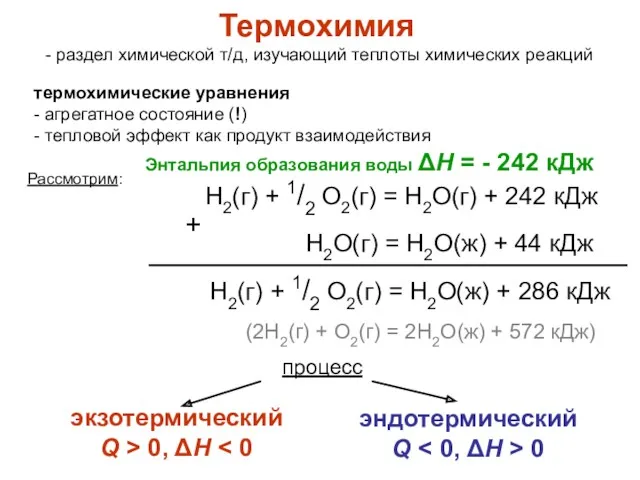
Термохимия
- раздел химической т/д, изучающий теплоты химических реакций
термохимические уравнения
- агрегатное
Термохимия
- раздел химической т/д, изучающий теплоты химических реакций
термохимические уравнения
- агрегатное

При проведении реакции в изобарных условиях тепловой эффект реакции определяется изменением
При проведении реакции в изобарных условиях тепловой эффект реакции определяется изменением

Тепловые эффекты можно рассчитывать!
Закон Гесса (II закон термохимии – также частный
Тепловые эффекты можно рассчитывать!
Закон Гесса (II закон термохимии – также частный

ΔН(1) – энтальпия образования CO (г)
ΔН(2) – энтальпия образования CO2 (г)
Следствие
ΔН(1) – энтальпия образования CO (г)
ΔН(2) – энтальпия образования CO2 (г)
Следствие

Стандартная энтальпия образования простых веществ
принята равной нулю
Хлор – газообразный, Cl2
Сера
Стандартная энтальпия образования простых веществ
принята равной нулю
Хлор – газообразный, Cl2
Сера
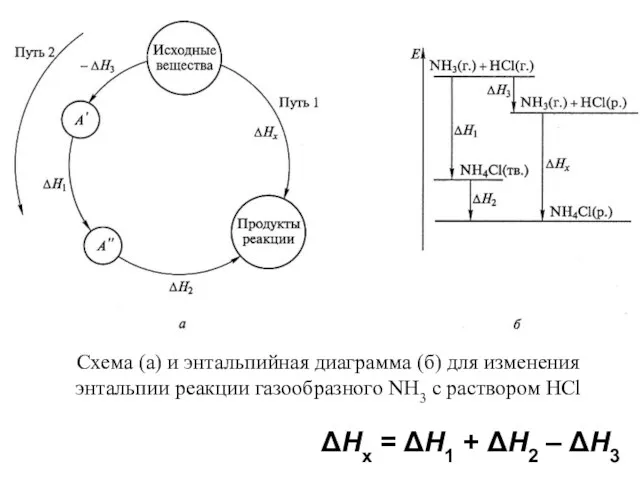
Схема (а) и энтальпийная диаграмма (б) для изменения энтальпии реакции газообразного
Схема (а) и энтальпийная диаграмма (б) для изменения энтальпии реакции газообразного

Энергия связи
-энергия, которую необходимо затратить на разрыв связи А-В
ΔН0A-B -энтальпия образования
Энергия связи
-энергия, которую необходимо затратить на разрыв связи А-В
ΔН0A-B -энтальпия образования

Атомарная теплота образования (Δa.f.Н0ABn)
-энтальпия образования данного вещества ABn из атомов
ABn =
Атомарная теплота образования (Δa.f.Н0ABn)
-энтальпия образования данного вещества ABn из атомов
ABn =
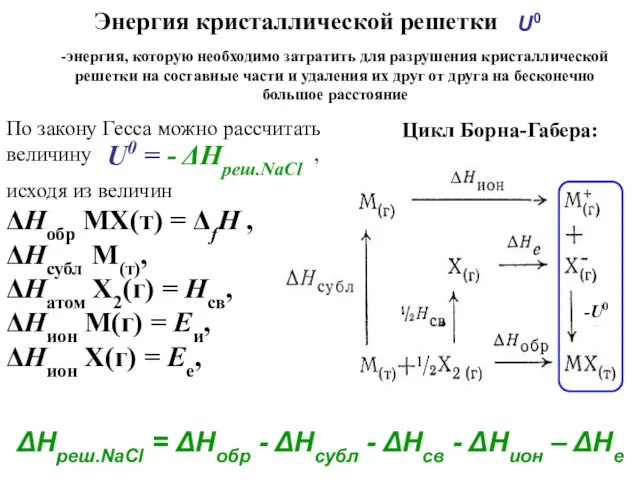
ΔНреш.NaCl = ΔНобр - ΔНсубл - ΔНсв - ΔНион – ΔНе
Цикл
ΔНреш.NaCl = ΔНобр - ΔНсубл - ΔНсв - ΔНион – ΔНе
Цикл
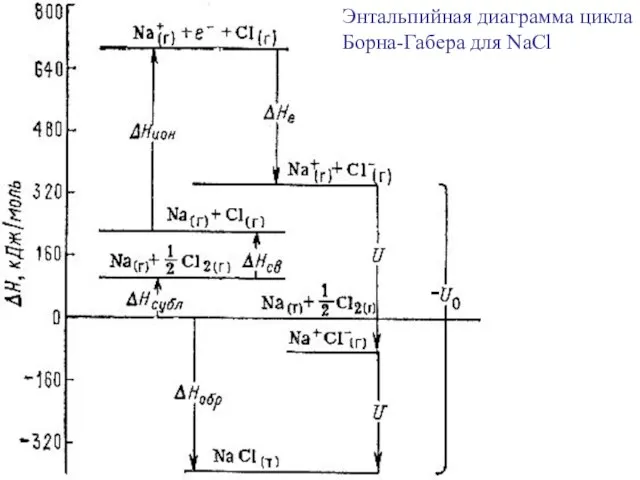
Энтальпийная диаграмма цикла Борна-Габера для NaCl
Энтальпийная диаграмма цикла Борна-Габера для NaCl
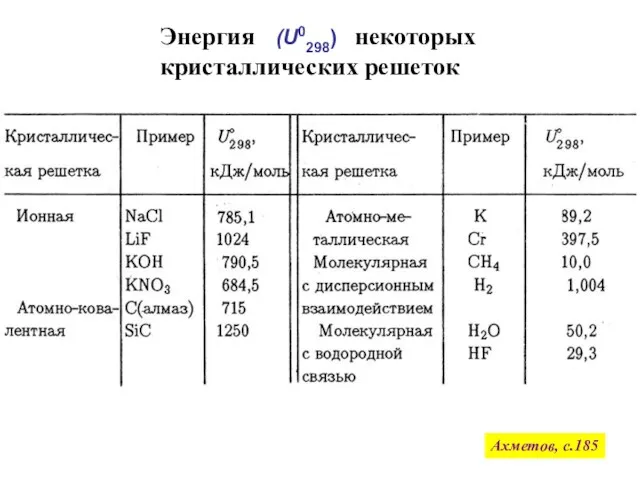
Энергия некоторых
кристаллических решеток
Ахметов, с.185
(U0298)
Энергия некоторых
кристаллических решеток
Ахметов, с.185
(U0298)

Изменением энтальпии можно охарактеризовать многие процессы
Энтальпия (теплота) сгорания (combustion)
- важная
Изменением энтальпии можно охарактеризовать многие процессы
Энтальпия (теплота) сгорания (combustion)
- важная

3. Процессы в растворах
Термохимические расчеты в растворах проводят не по теплотам
3. Процессы в растворах
Термохимические расчеты в растворах проводят не по теплотам

Для H+(aq), исходя из ΔfН0(298) = 0,
найдено ΔhН0(298) = -1075
Для H+(aq), исходя из ΔfН0(298) = 0,
найдено ΔhН0(298) = -1075

Растворение ионного соединения состоит из двух стадий – разрушения кристаллической решетки
Растворение ионного соединения состоит из двух стадий – разрушения кристаллической решетки

Выше для расчета энтальпии реакции использовали стандартные энтальпии образования продуктов и
Выше для расчета энтальпии реакции использовали стандартные энтальпии образования продуктов и

Кроме того, энтальпию сгорания можно использовать для расчета стандартной энтальпии образования
Кроме того, энтальпию сгорания можно использовать для расчета стандартной энтальпии образования

по энергиям связи (DA-B)
(например, когда неизвестна теплота образования одного из реагентов)
Результат
по энергиям связи (DA-B)
(например, когда неизвестна теплота образования одного из реагентов)
Результат

Температурная зависимость энтальпии
До сих пор рассматривались процессы, где температура исходных веществ
Температурная зависимость энтальпии
До сих пор рассматривались процессы, где температура исходных веществ
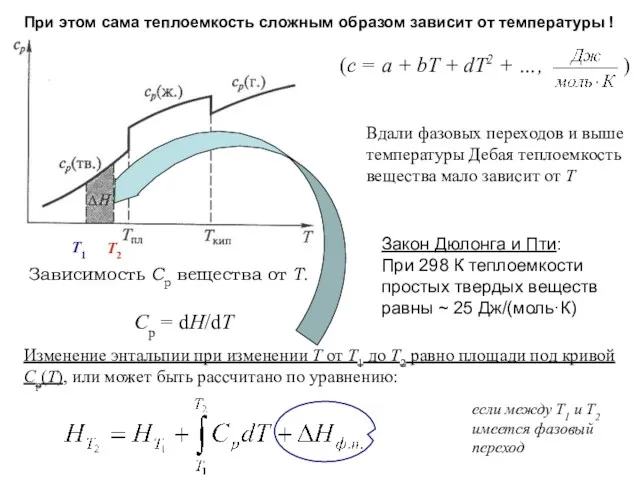
При этом сама теплоемкость сложным образом зависит от температуры !
Зависимость Ср
При этом сама теплоемкость сложным образом зависит от температуры !
Зависимость Ср
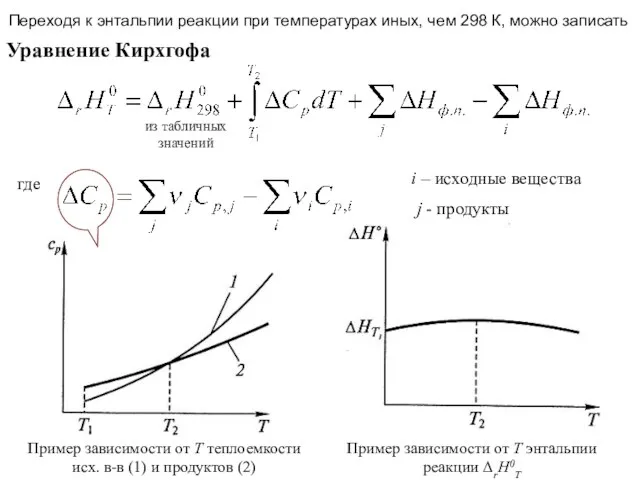
Пример зависимости от Т теплоемкости исх. в-в (1) и продуктов (2)
Пример
Пример зависимости от Т теплоемкости исх. в-в (1) и продуктов (2)
Пример

Если изучаемый температурный интервал невелик, и в нем не происходит фазовый
Если изучаемый температурный интервал невелик, и в нем не происходит фазовый

Направления процессов
в физико-химических системах
До XIX века полагали, что вещества реагируют,
Направления процессов
в физико-химических системах
До XIX века полагали, что вещества реагируют,

Мы уже можем рассчитать ΔrH, не прибегая к эксперименту.
А можно ли
Мы уже можем рассчитать ΔrH, не прибегая к эксперименту.
А можно ли

Sº(298)
растет при переходе тв – ж – г
в аморфном состоянии выше,
Sº(298)
растет при переходе тв – ж – г
в аморфном состоянии выше,
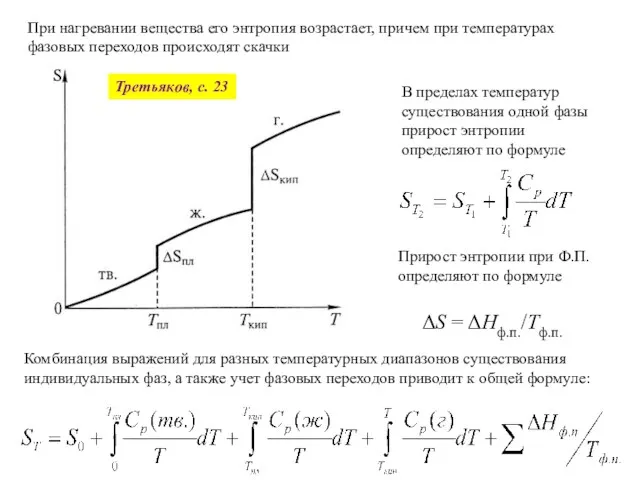
При нагревании вещества его энтропия возрастает, причем при температурах фазовых переходов
При нагревании вещества его энтропия возрастает, причем при температурах фазовых переходов

По данным о стандартной энтропии вещества можно рассчитать изменение энтропии химического
По данным о стандартной энтропии вещества можно рассчитать изменение энтропии химического

Приложение функций состояния к установлению направления процесса
Изменение энтальпии отражает стремление системы
Приложение функций состояния к установлению направления процесса
Изменение энтальпии отражает стремление системы

Формулировка второго начала термодинамики для неизолированных систем:
Невозможно осуществить перенос тепла от
Формулировка второго начала термодинамики для неизолированных систем:
Невозможно осуществить перенос тепла от

Возрастание энтропии в системе – «энтропийный фактор».
Количественно оценивают в виде произведения
TΔS
Возрастание энтропии в системе – «энтропийный фактор».
Количественно оценивают в виде произведения
TΔS

В чем состоит физический смысл энтропии?
Больцман (1896 г.) предложил определить это
В чем состоит физический смысл энтропии?
Больцман (1896 г.) предложил определить это

G - энергия Гиббса (свободная энергия)
- характеристика устойчивости системы при постоянном
G - энергия Гиббса (свободная энергия)
- характеристика устойчивости системы при постоянном
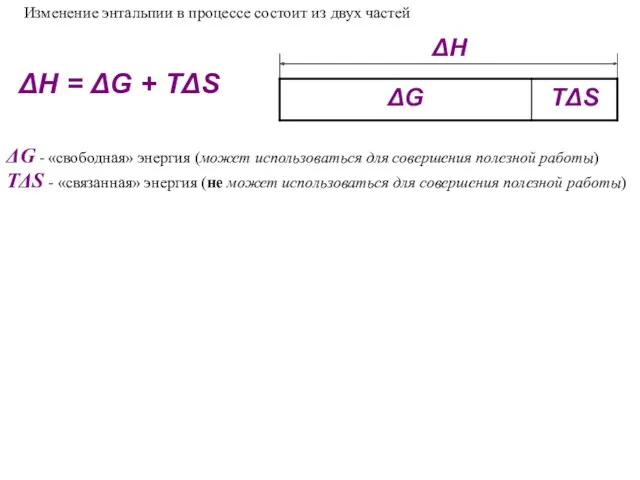
ΔH = ΔG + TΔS
Изменение энтальпии в процессе состоит из двух
ΔH = ΔG + TΔS
Изменение энтальпии в процессе состоит из двух

ΔG = (ΔН – TΔS) < 0
Для самопроизвольного протекания процесса при
ΔG = (ΔН – TΔS) < 0
Для самопроизвольного протекания процесса при

1
Для практических целей используют значения энергии Гиббса реакций для стандартных условий
1
Для практических целей используют значения энергии Гиббса реакций для стандартных условий

Вещества, для которых ΔG0298 < 0, являются термодинамически стабильными, следовательно, если
Вещества, для которых ΔG0298 < 0, являются термодинамически стабильными, следовательно, если

Третий путь определения стандартного изменения энергии Гиббса реакции основан на комбинации
Третий путь определения стандартного изменения энергии Гиббса реакции основан на комбинации

Фримантл, с. 254
Диаграммы Эллингема
позволяют наглядно определять, какой оксид будет восстанавливаться в
Фримантл, с. 254
Диаграммы Эллингема
позволяют наглядно определять, какой оксид будет восстанавливаться в
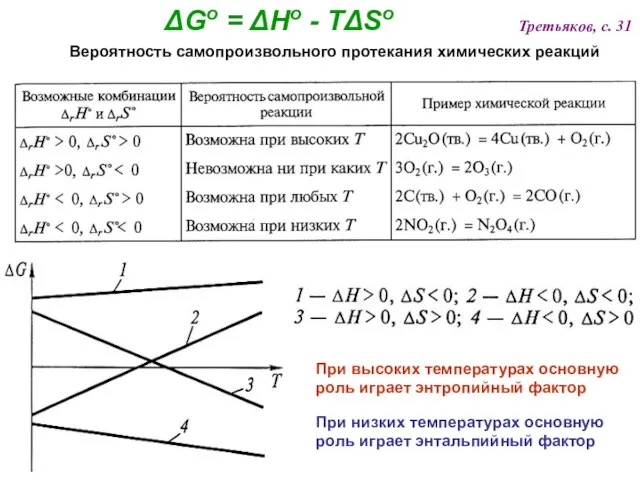
Третьяков, с. 31
Вероятность самопроизвольного протекания химических реакций
При высоких температурах основную роль
Третьяков, с. 31
Вероятность самопроизвольного протекания химических реакций
При высоких температурах основную роль

Итак, многие химические процессы, начиная протекать в одном направлении, затем идут
Итак, многие химические процессы, начиная протекать в одном направлении, затем идут

В обратимом процессе через некоторое время устанавливается состояние равновесия
При постоянных внешних
В обратимом процессе через некоторое время устанавливается состояние равновесия
При постоянных внешних

Кажущееся равновесие - заторможенное (метастабильное состояние)
К.р. сходно с Истинным р. по
Кажущееся равновесие - заторможенное (метастабильное состояние)
К.р. сходно с Истинным р. по

Тогда для гомогенной реакции
справедливо:
aA + bB + … ⮀ dD +
Тогда для гомогенной реакции
справедливо:
aA + bB + … ⮀ dD +
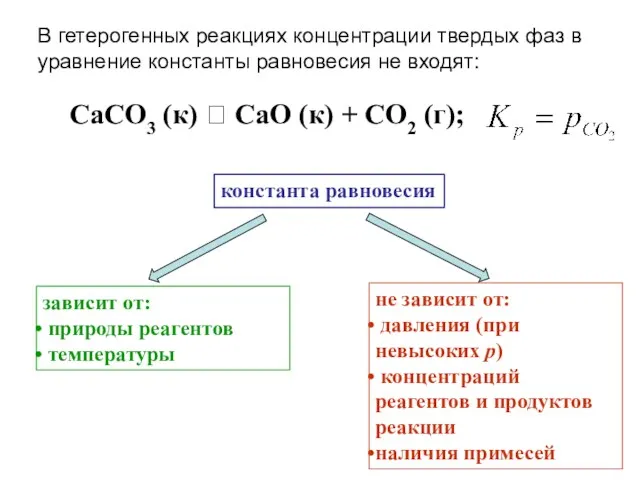
В гетерогенных реакциях концентрации твердых фаз в уравнение константы равновесия не
В гетерогенных реакциях концентрации твердых фаз в уравнение константы равновесия не
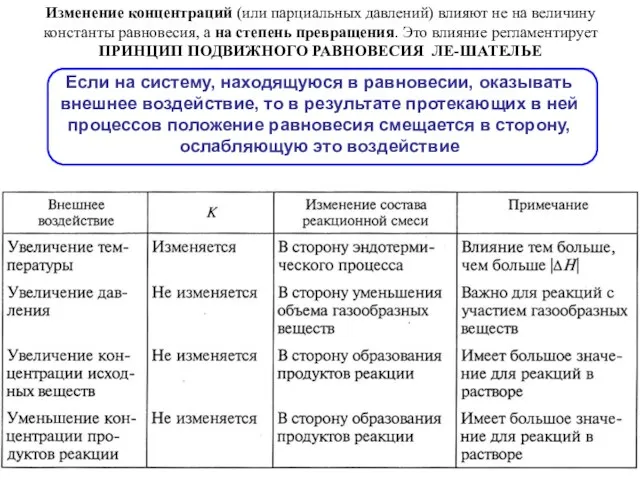
Изменение концентраций (или парциальных давлений) влияют не на величину константы равновесия,
Изменение концентраций (или парциальных давлений) влияют не на величину константы равновесия,
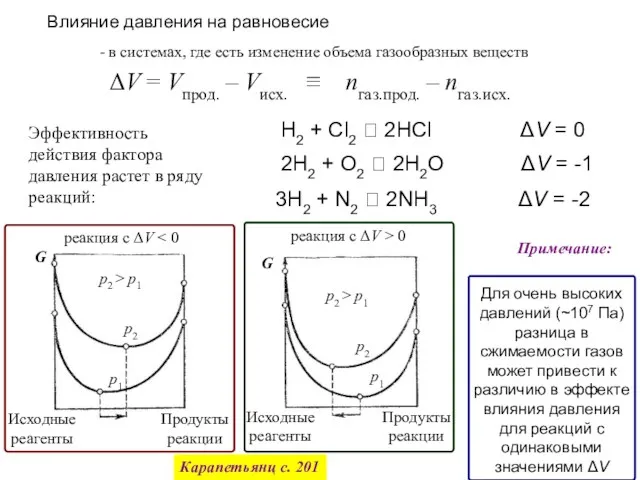
Влияние давления на равновесие
- в системах, где есть изменение объема газообразных
Влияние давления на равновесие
- в системах, где есть изменение объема газообразных

Влияние концентрации на равновесие
В соответствии с принципом Ле-Шателье введение в равновесную
Влияние концентрации на равновесие
В соответствии с принципом Ле-Шателье введение в равновесную

Термодинамика дает важное соотношение (изотерма реакции):
Изменения ΔG в реальных условиях химических
Термодинамика дает важное соотношение (изотерма реакции):
Изменения ΔG в реальных условиях химических
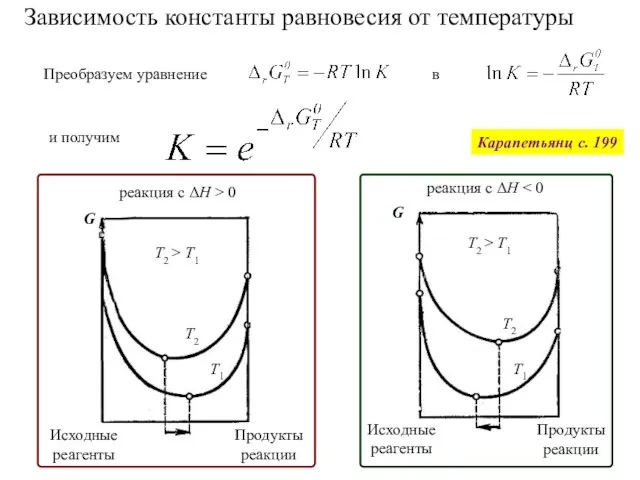
Зависимость константы равновесия от температуры
Преобразуем уравнение
в
и получим
Карапетьянц с. 199
Зависимость константы равновесия от температуры
Преобразуем уравнение
в
и получим
Карапетьянц с. 199
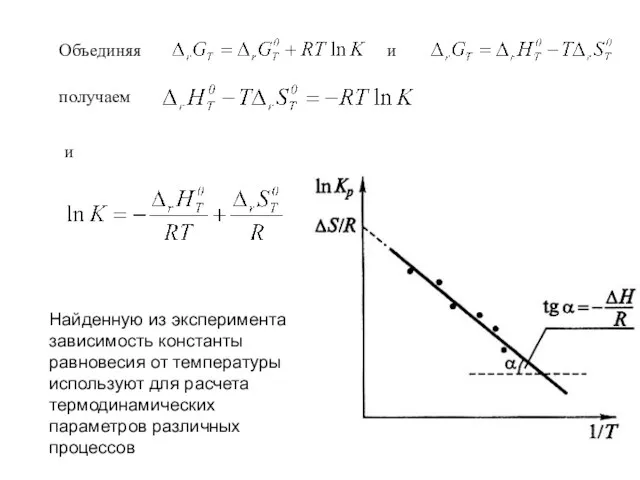
Объединяя
и
получаем
и
Найденную из эксперимента зависимость константы равновесия от температуры используют для расчета
Объединяя
и
получаем
и
Найденную из эксперимента зависимость константы равновесия от температуры используют для расчета

Скорость и механизм химических реакций
Как мы выяснили, реакции с ΔG >
Скорость и механизм химических реакций
Как мы выяснили, реакции с ΔG >
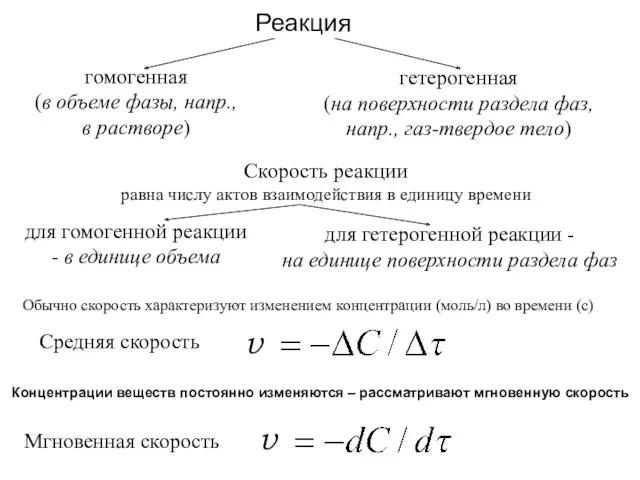
Реакция
гомогенная
(в объеме фазы, напр., в растворе)
гетерогенная
(на поверхности раздела фаз,
Реакция
гомогенная
(в объеме фазы, напр., в растворе)
гетерогенная
(на поверхности раздела фаз,
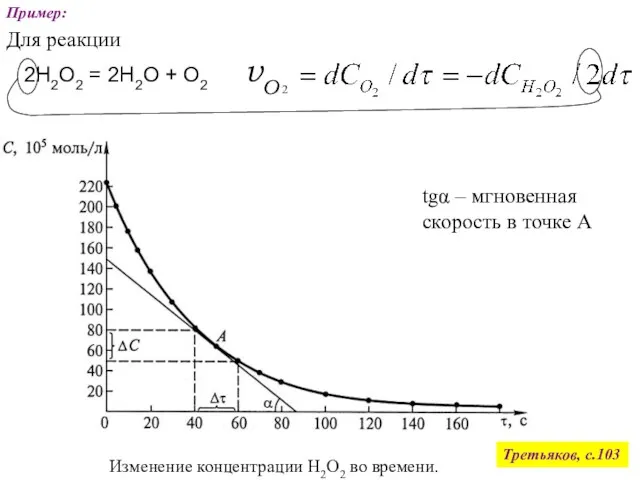
Пример:
Третьяков, с.103
Для реакции
2Н2О2 = 2Н2О + О2
Изменение концентрации Н2О2 во времени.
tgα
Пример:
Третьяков, с.103
Для реакции
2Н2О2 = 2Н2О + О2
Изменение концентрации Н2О2 во времени.
tgα

Скорость химической реакции зависит от многих факторов:
природы реагирующих веществ
концентрации реагирующих веществ
температуры
наличия
Скорость химической реакции зависит от многих факторов:
природы реагирующих веществ
концентрации реагирующих веществ
температуры
наличия

m и n равны стехиометрическим коэффициентам только в том случае, если
m и n равны стехиометрическим коэффициентам только в том случае, если
![Для реакции разложения Н2О2: v = k[Н2О2]1 Кинетические уравнения для](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/415613/slide-64.jpg)
Для реакции разложения Н2О2:
v = k[Н2О2]1
Кинетические уравнения для реакций с различным
Для реакции разложения Н2О2:
v = k[Н2О2]1
Кинетические уравнения для реакций с различным

На практике порядок реакции по реагенту определяют из графика в логарифмических
На практике порядок реакции по реагенту определяют из графика в логарифмических

Физический смысл величины k:
константа скорости реакции численно равна скорости реакции при
Физический смысл величины k:
константа скорости реакции численно равна скорости реакции при

Период полупревращения вещества
- время, за которое прореагирует половина его количества
Интегрированием
Период полупревращения вещества
- время, за которое прореагирует половина его количества
Интегрированием

Итак:
Только элементарные реакции идут так, как они записаны, сложные реакции (большинство)
Итак:
Только элементарные реакции идут так, как они записаны, сложные реакции (большинство)

«Кинетический» вывод константы равновесия
Для обратимой одностадийной реакции
А + В ⮀
«Кинетический» вывод константы равновесия
Для обратимой одностадийной реакции
А + В ⮀
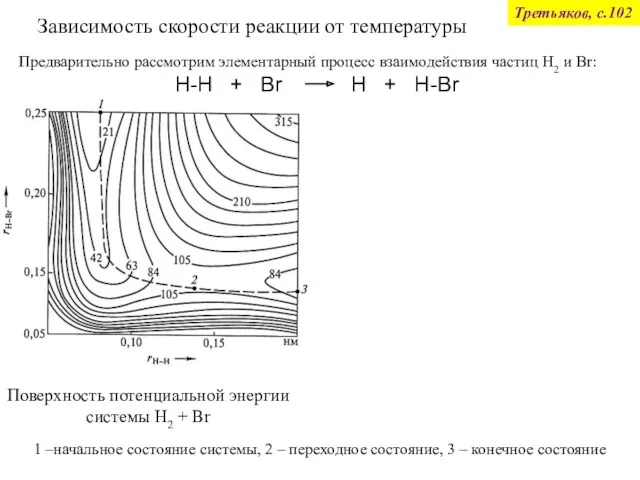
Зависимость скорости реакции от температуры
Третьяков, с.102
Поверхность потенциальной энергии
системы H2 + Br
Профиль
Зависимость скорости реакции от температуры
Третьяков, с.102
Поверхность потенциальной энергии
системы H2 + Br
Профиль

Зависимость скорости реакции от температуры
В подавляющем большинстве случаев скорость реакции с
Зависимость скорости реакции от температуры
В подавляющем большинстве случаев скорость реакции с

«Температура ускоряет реакции, потому что молекулы чаще соударяются»
неверный ответ!
при ΔТ =
«Температура ускоряет реакции, потому что молекулы чаще соударяются»
неверный ответ!
при ΔТ =
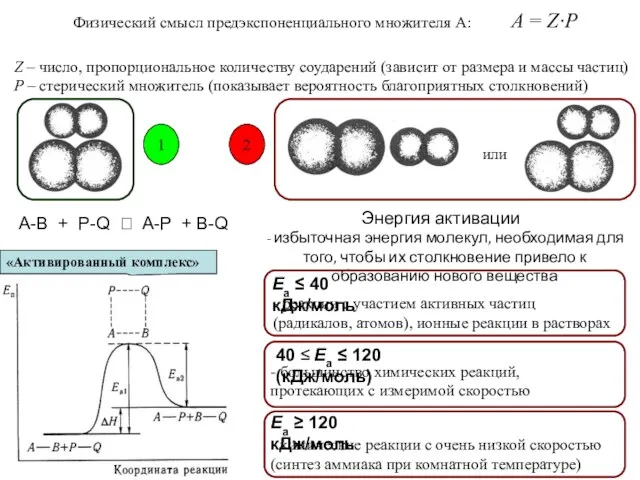
Физический смысл предэкспоненциального множителя А: A = Z·P
Z – число, пропорциональное
Физический смысл предэкспоненциального множителя А: A = Z·P
Z – число, пропорциональное
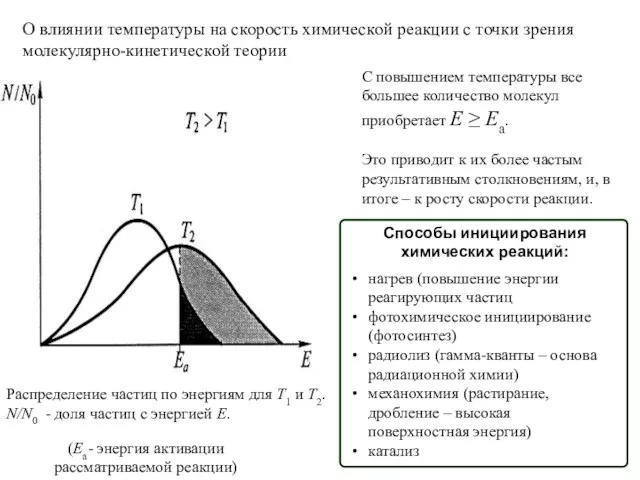
О влиянии температуры на скорость химической реакции с точки зрения молекулярно-кинетической
О влиянии температуры на скорость химической реакции с точки зрения молекулярно-кинетической
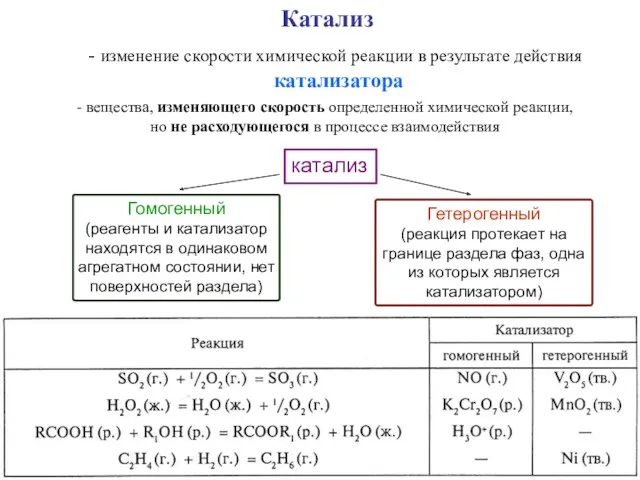
Катализ
изменение скорости химической реакции в результате действия
катализатора
- вещества, изменяющего скорость
Катализ
изменение скорости химической реакции в результате действия
катализатора
- вещества, изменяющего скорость
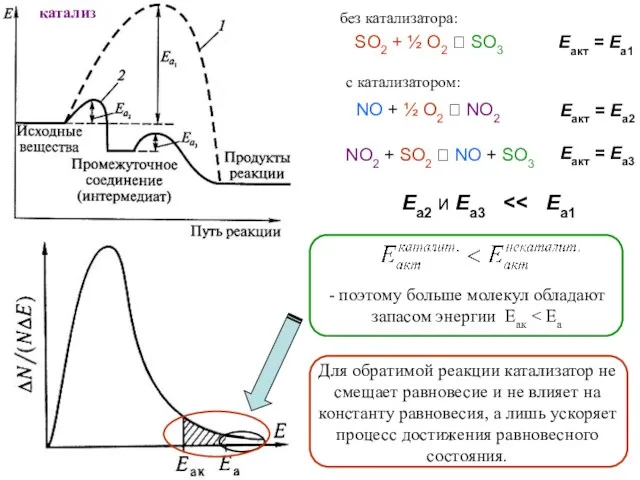
без катализатора:
с катализатором:
SO2 + ½ O2 ⮀ SO3
Eакт = Еа1
NO +
без катализатора:
с катализатором:
SO2 + ½ O2 ⮀ SO3
Eакт = Еа1
NO +
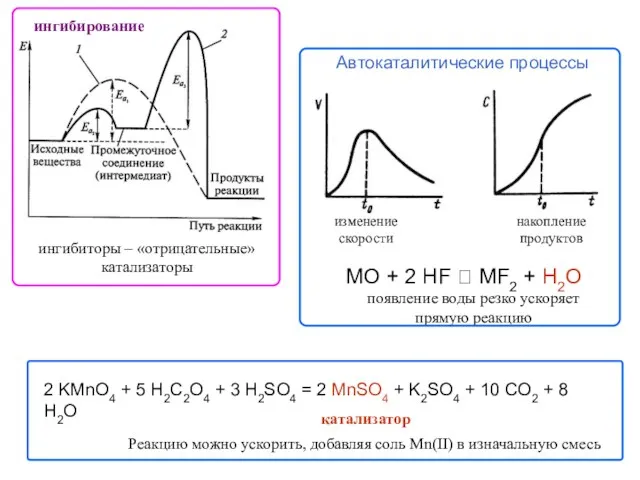
ингибирование
Автокаталитические процессы
MO + 2 HF ⮀ MF2 + H2O
появление воды резко
ингибирование
Автокаталитические процессы
MO + 2 HF ⮀ MF2 + H2O
появление воды резко

Гомогенный катализ
в газовой фазе – рассмотрен выше на примере реакции SO2
Гомогенный катализ
в газовой фазе – рассмотрен выше на примере реакции SO2

Гетерогенный катализ
Процессы идут на границе раздела фаз,
поэтому для объяснения привлекают
Гетерогенный катализ
Процессы идут на границе раздела фаз,
поэтому для объяснения привлекают
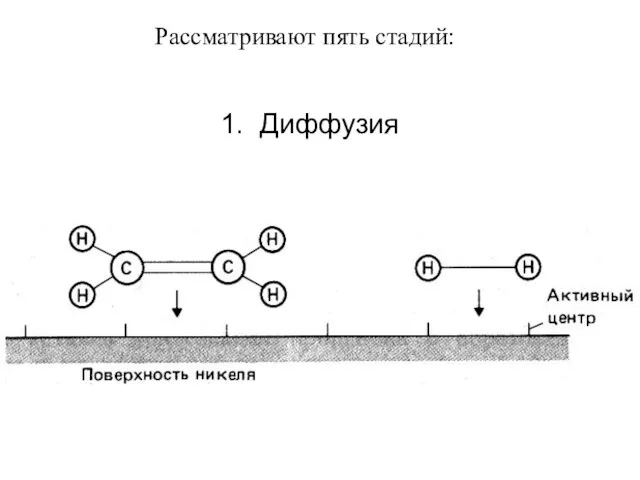
Рассматривают пять стадий:
1. Диффузия
Рассматривают пять стадий:
1. Диффузия
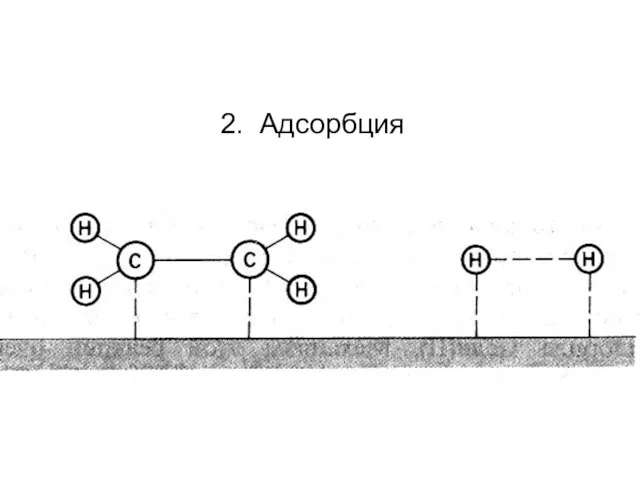
2. Адсорбция
2. Адсорбция
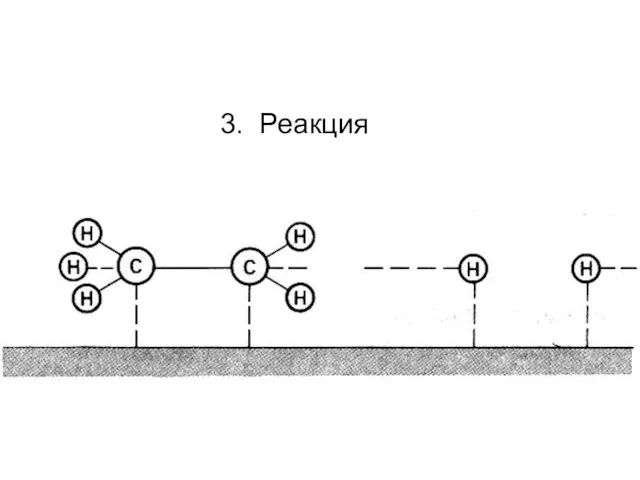
3. Реакция
3. Реакция
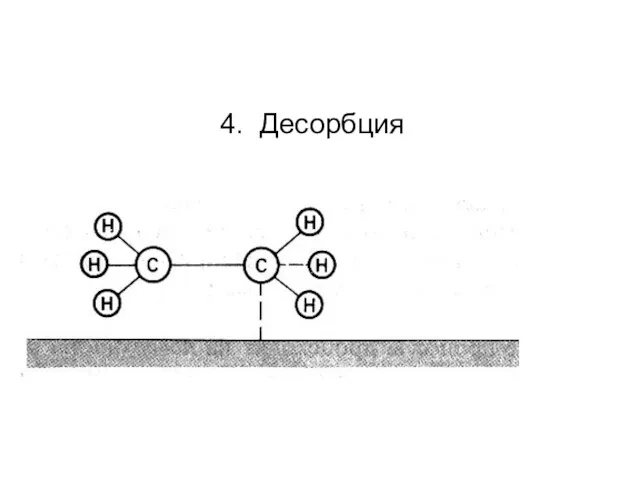
4. Десорбция
4. Десорбция
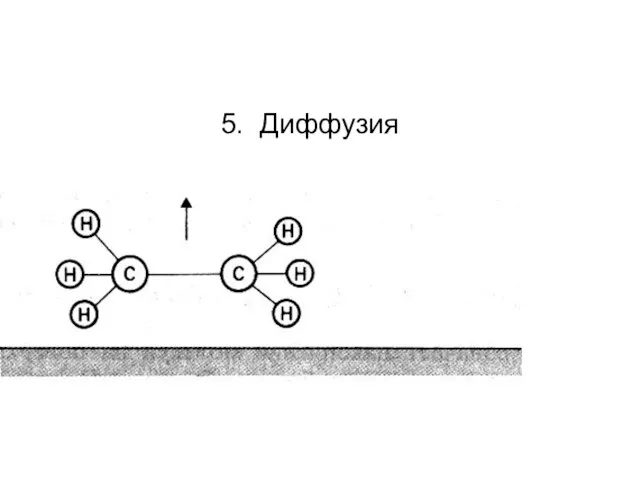
5. Диффузия
5. Диффузия
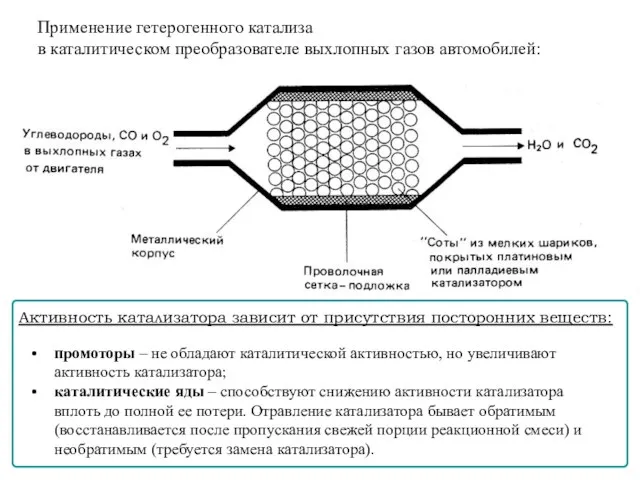
Активность катализатора зависит от присутствия посторонних веществ:
промоторы – не обладают каталитической
Активность катализатора зависит от присутствия посторонних веществ:
промоторы – не обладают каталитической
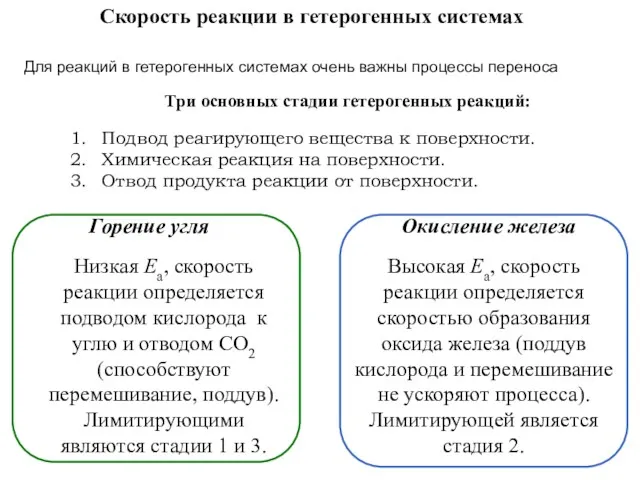
Скорость реакции в гетерогенных системах
Для реакций в гетерогенных системах очень важны
Скорость реакции в гетерогенных системах
Для реакций в гетерогенных системах очень важны
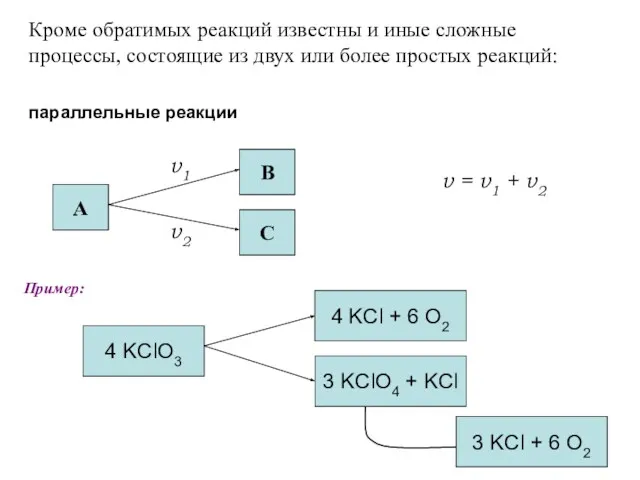
Кроме обратимых реакций известны и иные сложные процессы, состоящие из двух
Кроме обратимых реакций известны и иные сложные процессы, состоящие из двух
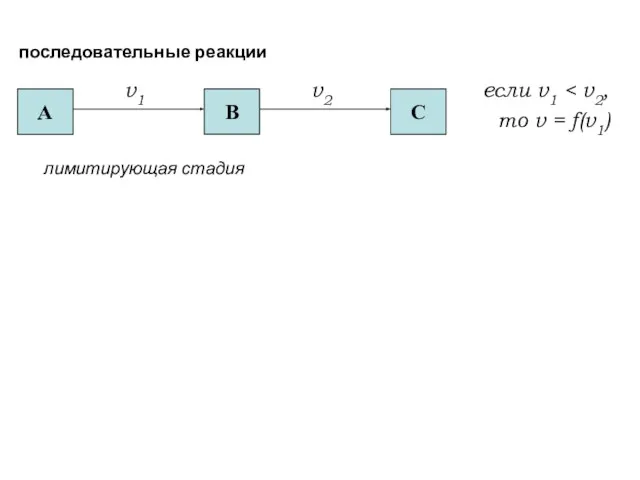
если v1 < v2,
то v = f(v1)
лимитирующая стадия
H2O2
2 OH*
OH* + HI
Пример:
I
если v1 < v2,
то v = f(v1)
лимитирующая стадия
H2O2
2 OH*
OH* + HI
Пример:
I
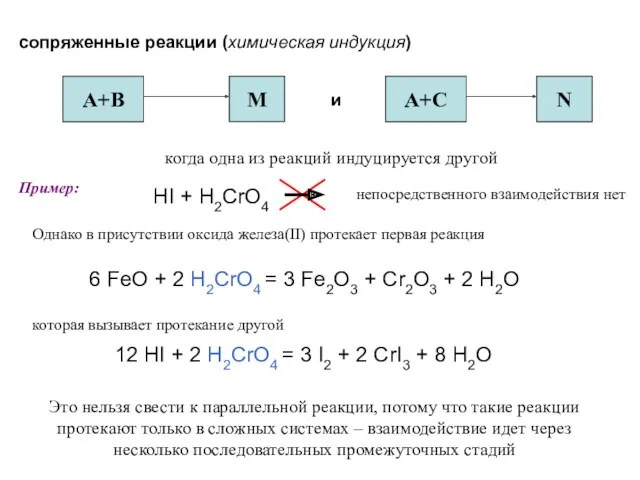
сопряженные реакции (химическая индукция)
А+В
M
N
А+C
и
когда одна из реакций индуцируется другой
Пример:
HI + H2CrO4
6
сопряженные реакции (химическая индукция)
А+В
M
N
А+C
и
когда одна из реакций индуцируется другой
Пример:
HI + H2CrO4
6
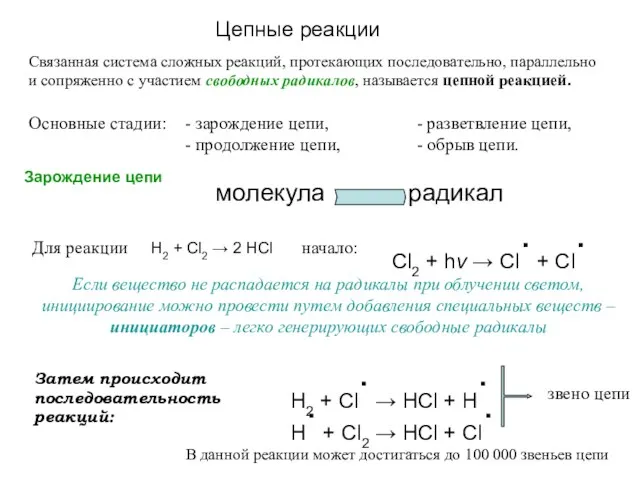
Цепные реакции
Связанная система сложных реакций, протекающих последовательно, параллельно и сопряженно с
Цепные реакции
Связанная система сложных реакций, протекающих последовательно, параллельно и сопряженно с
 Высокомолекулярные соединения и супрамолекулярные структуры. (Лекция 3)
Высокомолекулярные соединения и супрамолекулярные структуры. (Лекция 3) Кислородсодержащие соединения серы. Оксиды серы
Кислородсодержащие соединения серы. Оксиды серы Табиғи нанокристалдар
Табиғи нанокристалдар Кислоты 8 класс
Кислоты 8 класс Летучие яды
Летучие яды Тепловой эффект химических реакций
Тепловой эффект химических реакций Химическая связь. 8 класс
Химическая связь. 8 класс Товары бытовой химии
Товары бытовой химии Химическая промышленность
Химическая промышленность Топливо. Виды топлива
Топливо. Виды топлива Тканые армирующие наполнители
Тканые армирующие наполнители Конденсация Кляйзена
Конденсация Кляйзена Нефтепродукты. Продукты переработки нефти
Нефтепродукты. Продукты переработки нефти Химическая термодинамика
Химическая термодинамика Как и где используется соляная кислота
Как и где используется соляная кислота Аммиак
Аммиак Элементы подгруппы углерода
Элементы подгруппы углерода Применение жиров
Применение жиров А.М. Бутлеровтың химиялык кұрылыс теориясы
А.М. Бутлеровтың химиялык кұрылыс теориясы Строение вещества и агрегатные состояния вещества
Строение вещества и агрегатные состояния вещества Свойства, состав и применение пластмасс
Свойства, состав и применение пластмасс Полисахариды. Крахмал
Полисахариды. Крахмал Технологии получения полимерных нанокомпозитов
Технологии получения полимерных нанокомпозитов Оксиды
Оксиды Алкалоиды: распространение в природе, получение, применение, способы синтеза
Алкалоиды: распространение в природе, получение, применение, способы синтеза Электролиз
Электролиз Возникновение и развитие органической химии. Теория химического строения. Структурные формулы
Возникновение и развитие органической химии. Теория химического строения. Структурные формулы Цинк
Цинк