- Генетика. Синдром марфана (Marfan syndrom)

Содержание
- 2. Минимальные диагностические признаки: - высокий рост - арахнодактилия - гиперподвижность суставов - подвывих хрусталика - расширение
- 3. Характерно поражение трех систем органов: - скелет - глаза - сердечно-сосудистая система
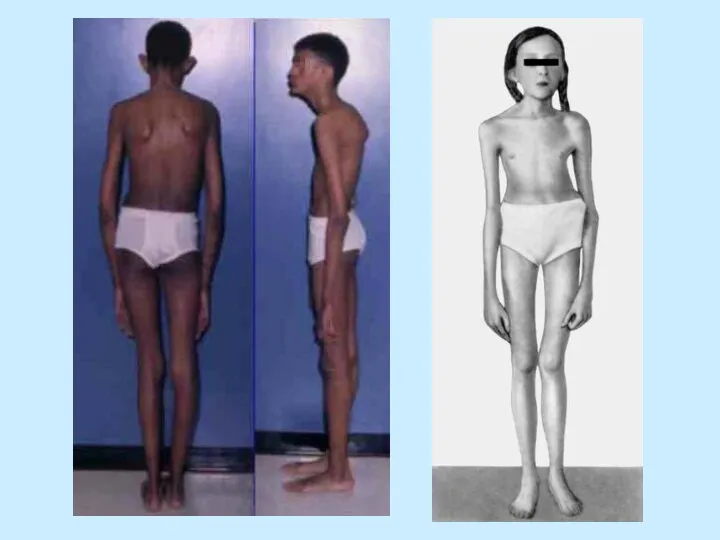
- 4. ВЫСОКИЙ РОСТ Средняя длина при рождении у мальчиков – 53 см + / - 4,4 см
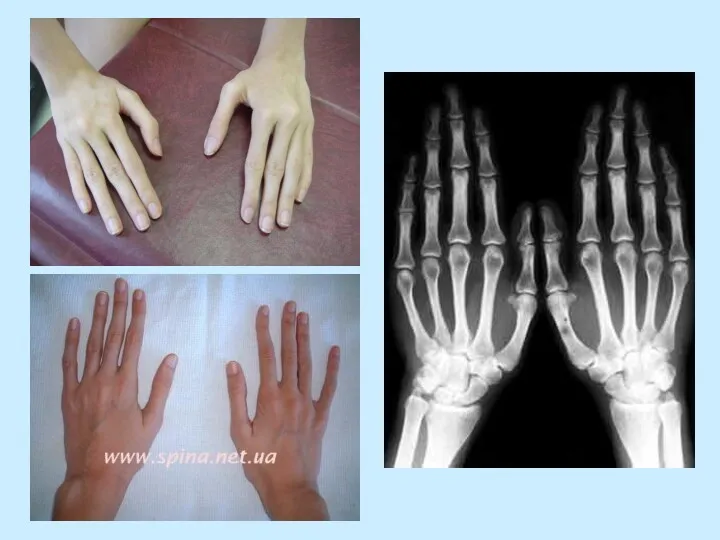
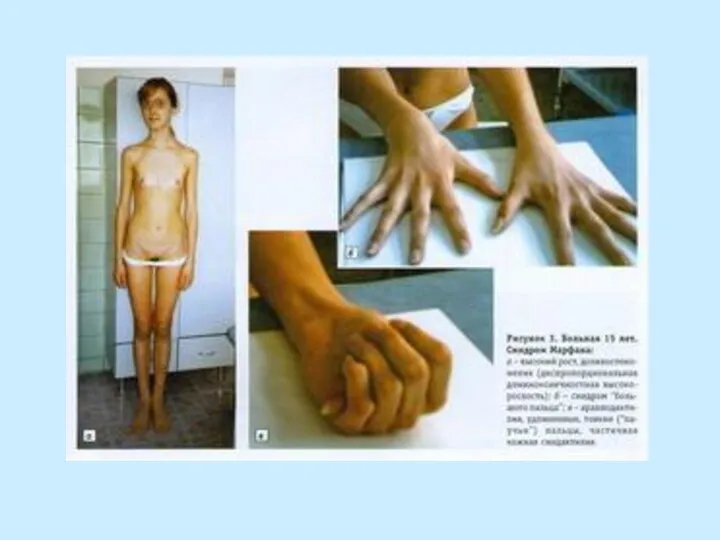
- 5. СКЕЛЕТ: - конечности длинные и тонкие (особенно дистальные отделы) - размах рук больше роста - арахнодактилия
- 6. - суставы гипермобильные - контрактуры суставов - сколиоз (чаще правосторонний) - 60% - кифоз - воронкообразная,
- 7. ГЛАЗА: - двусторонний подвывих хрусталика – 75% - иридодонез (дрожание радужки) - гипоплазия радужки - сферофакия
- 8. СЕРДЕЧНО-СОСУДИСТАЯ СИСТЕМА - расширение восходящей части аорты - пролапс митрального клапана - пролапс трехстворчатого клапана -
- 9. ПОРАЖЕНИЕ ДРУГИХ СИСТЕМ: - ранний артрит - бедренные, паховые, диафрагмальные грыжи - атрофические стрии - мышечная
- 10. При КТ и МРТ у большинства больных находят эктазию твердой оболочки люмбо-сакрального отдела спинного мозга. Во
- 11. Основные критерии диагноза по Берлинской классификации (1988 год): 1) при отсутствии пораженного родственника I степени родства
- 12. Гентская классификация (1996 год) Выделяют 4 главных критерия: - расширение (расслоение) корня аорты - подвывих хрусталика
- 13. - Тип наследования – аутосомно-доминантный с полной пенетрантностью и варьирующей экспрессивностью - 75% всех случаев –
- 14. Мутации в гене FBN1 обуславливают и другие фенотипы, включающие отдельные проявления синдрома Марфана: - синдром пролапса
- 15. Синдром Марфана 2 - OMIM: 154705 - Синоним: Марфаноподобное поражение соединительной ткани - Клиника: скелетные и
- 16. Дифференциальный диагноз: - гомоцистинурия - контрактурная врожденная арахнодактилия - наследственная прогрессирующая артроофтальмопатия - синдром Марфана 2
- 23. Скачать презентацию

Минимальные диагностические признаки:
- высокий рост
- арахнодактилия
- гиперподвижность суставов
- подвывих хрусталика
- расширение
Минимальные диагностические признаки: - высокий рост - арахнодактилия - гиперподвижность суставов - подвывих хрусталика - расширение

Характерно поражение трех систем органов:
- скелет
- глаза
- сердечно-сосудистая система
Характерно поражение трех систем органов:
- скелет
- глаза
- сердечно-сосудистая система

ВЫСОКИЙ РОСТ
Средняя длина при рождении у мальчиков – 53 см
+
ВЫСОКИЙ РОСТ Средняя длина при рождении у мальчиков – 53 см +

СКЕЛЕТ:
- конечности длинные и тонкие (особенно дистальные отделы)
- размах рук больше
СКЕЛЕТ:
- конечности длинные и тонкие (особенно дистальные отделы)
- размах рук больше

- суставы гипермобильные
- контрактуры суставов
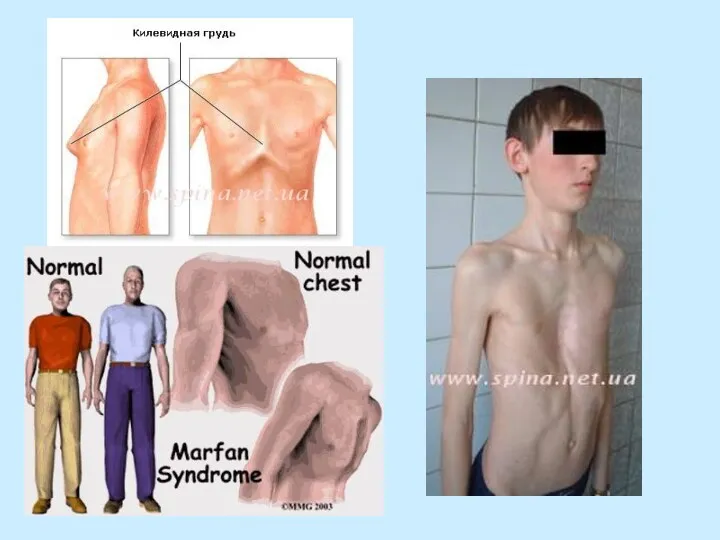
- сколиоз (чаще правосторонний) - 60%
- кифоз
- воронкообразная, килевидная грудная клетка
- плоскостопие
- долихоцефалия
- высокое аркообразное
- суставы гипермобильные - контрактуры суставов - сколиоз (чаще правосторонний) - 60% - кифоз - воронкообразная, килевидная грудная клетка - плоскостопие - долихоцефалия - высокое аркообразное

ГЛАЗА:
- двусторонний подвывих хрусталика – 75%
- иридодонез (дрожание радужки)
- гипоплазия радужки
-
ГЛАЗА: - двусторонний подвывих хрусталика – 75% - иридодонез (дрожание радужки) - гипоплазия радужки -

СЕРДЕЧНО-СОСУДИСТАЯ СИСТЕМА
- расширение восходящей части аорты
- пролапс митрального клапана
- пролапс трехстворчатого
СЕРДЕЧНО-СОСУДИСТАЯ СИСТЕМА - расширение восходящей части аорты - пролапс митрального клапана - пролапс трехстворчатого

ПОРАЖЕНИЕ ДРУГИХ СИСТЕМ:
- ранний артрит
- бедренные, паховые, диафрагмальные грыжи
- атрофические стрии
-
ПОРАЖЕНИЕ ДРУГИХ СИСТЕМ: - ранний артрит - бедренные, паховые, диафрагмальные грыжи - атрофические стрии -

При КТ и МРТ у большинства больных находят эктазию твердой оболочки
При КТ и МРТ у большинства больных находят эктазию твердой оболочки

Основные критерии диагноза по Берлинской классификации (1988 год):
1) при отсутствии пораженного
Основные критерии диагноза по Берлинской классификации (1988 год): 1) при отсутствии пораженного

Гентская классификация (1996 год)
Выделяют 4 главных критерия:
- расширение (расслоение) корня аорты
-
Гентская классификация (1996 год) Выделяют 4 главных критерия: - расширение (расслоение) корня аорты -

- Тип наследования – аутосомно-доминантный с полной пенетрантностью и варьирующей экспрессивностью
-
- Тип наследования – аутосомно-доминантный с полной пенетрантностью и варьирующей экспрессивностью -

Мутации в гене FBN1 обуславливают и другие фенотипы, включающие отдельные проявления
Мутации в гене FBN1 обуславливают и другие фенотипы, включающие отдельные проявления

Синдром Марфана 2
- OMIM: 154705
- Синоним: Марфаноподобное поражение соединительной ткани
- Клиника:
Синдром Марфана 2 - OMIM: 154705 - Синоним: Марфаноподобное поражение соединительной ткани - Клиника:

Дифференциальный диагноз:
- гомоцистинурия
- контрактурная врожденная арахнодактилия
- наследственная прогрессирующая артроофтальмопатия
- синдром
Дифференциальный диагноз: - гомоцистинурия - контрактурная врожденная арахнодактилия - наследственная прогрессирующая артроофтальмопатия - синдром

 Симптоматическое и климатическое бесплодие
Симптоматическое и климатическое бесплодие Сахарный диабет
Сахарный диабет Тениаринхоз. Возбудитель тениаринхоза. Осложнения. Лечение
Тениаринхоз. Возбудитель тениаринхоза. Осложнения. Лечение Болезни нервов у животных
Болезни нервов у животных Заболевания конъюнктивы
Заболевания конъюнктивы Харчові захворювання мікробного походження. Основні заходи їхньої профілактики
Харчові захворювання мікробного походження. Основні заходи їхньої профілактики Атом энергетикасы өндірісі мен басқа да радиологиялық нысандардағы апаттардың себебінен жіктелуі
Атом энергетикасы өндірісі мен басқа да радиологиялық нысандардағы апаттардың себебінен жіктелуі Біомедична етика: основні теоретичні та прикладні аспекти у сучасному суспільстві
Біомедична етика: основні теоретичні та прикладні аспекти у сучасному суспільстві Шетелдегі инклюзивті білім беру
Шетелдегі инклюзивті білім беру Артериальная гипертензия у детей и подростков (диагностика, лечение, профилактика)
Артериальная гипертензия у детей и подростков (диагностика, лечение, профилактика) Африканская чума свиней
Африканская чума свиней Болезни кишечника
Болезни кишечника Специальные знания в генетической экспертизе
Специальные знания в генетической экспертизе Вирус иммунодефицита человека (ВИЧ). Вирус гриппа
Вирус иммунодефицита человека (ВИЧ). Вирус гриппа Этика и деонтология в медицине
Этика и деонтология в медицине Эшерихии - кишечная палочка. Микробиология
Эшерихии - кишечная палочка. Микробиология Течение и ведение послеродового периода. Становление лактации. Послеродовая контрацепция
Течение и ведение послеродового периода. Становление лактации. Послеродовая контрацепция Гастроэзофагеальная рефлюксная болезнь
Гастроэзофагеальная рефлюксная болезнь Воспаление. Этиология воспаления
Воспаление. Этиология воспаления Снотворные, противоэпилептические и противопаркинсонические средства
Снотворные, противоэпилептические и противопаркинсонические средства Жизнь без наркотиков
Жизнь без наркотиков Рухова активність, як основа здорової поведінки людини
Рухова активність, як основа здорової поведінки людини Физиология крови
Физиология крови Бинокулярное зрение. Косоглазие
Бинокулярное зрение. Косоглазие Предмет и задачи патоморфологии. Методы патоморфологических исследований. Основные этапы развития патоморфологии
Предмет и задачи патоморфологии. Методы патоморфологических исследований. Основные этапы развития патоморфологии Мүгедектерге көрсетілетін әлеуметтік көмек түрлері
Мүгедектерге көрсетілетін әлеуметтік көмек түрлері Эпилепсия. Классификация эпилепсии. Диагностические критерии. Неотложная помощь. Принципы лечения
Эпилепсия. Классификация эпилепсии. Диагностические критерии. Неотложная помощь. Принципы лечения Физиология выделительной системы
Физиология выделительной системы