- Синдромы частичных анеуплоидий

Содержание
- 2. СИНДРОМ ТРИСОМИИ ПО КОРОТКОМУ ПЛЕЧУ 9-Й ХРОМОСОМЫ - наиболее частая форма частичных трисомий (опубликовано около 200
- 3. ЖЕНЩИНА 21 ГОД Ребенок 3-х лет
- 5. СИНДРОМ КОЛЬЦЕВОЙ ХРОМОСОМЫ 9 Кариотип 46 ХХ или ХУ, r (9). Основные клинико-морфологические признаки: характерное лицо
- 6. СИНДРОМ КОШАЧЬЕГО КРИКА - частичная моносомия по короткому плечу хромосомы 5 . Синдром моносомии 5р- был
- 7. Клиническая картина синдрома 5р- довольно сильно различается у отдельных больных по сочетанию врожденных пороков развития органов.
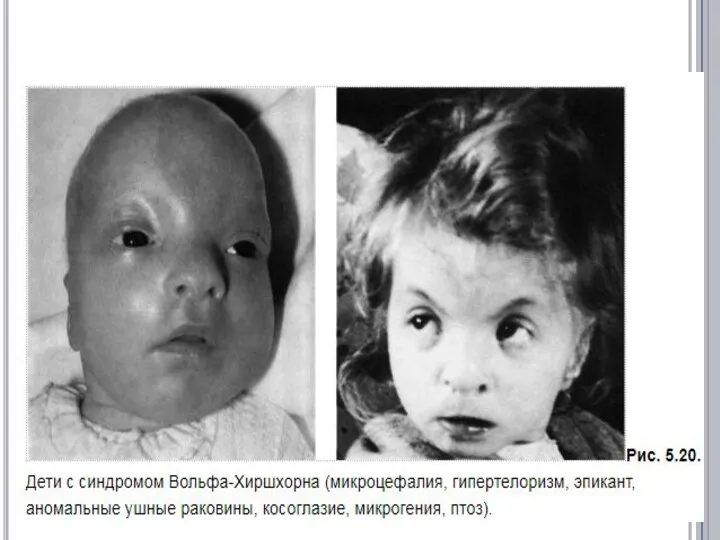
- 8. Ребенок с выраженными признаками синдрома «кошачьего крика» (микроцефалия, лунообразное лицо, эпикант, гипертелоризм, широкая плоская спинка носа,
- 9. СИНДРОМ ВОЛЬФА-ХИРШХОРНА Популяционная частота - 1:100000. обусловлен делецией сегмента короткого плеча хромосомы 4. Симптомы: задержка роста,
- 12. СИНДРОМ ОРБЕЛИ обусловлен делецией длинного плеча 13-й хромосомы. Популяционная частота не установлена. Характерны микроцефалия, отсутствие носовой
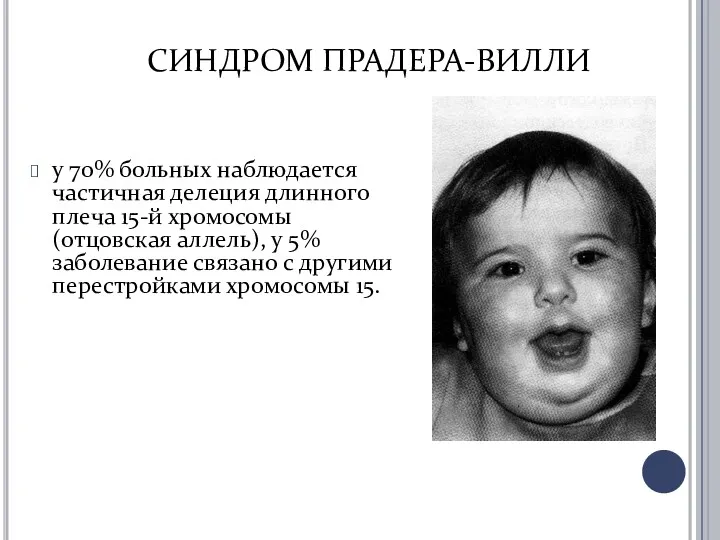
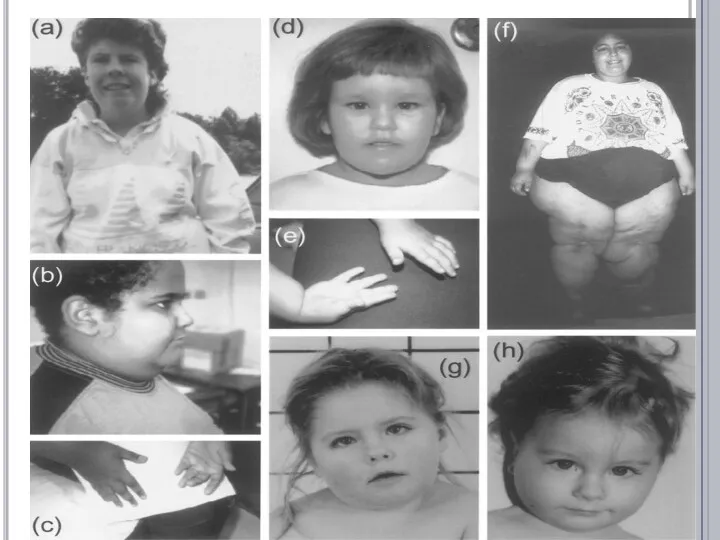
- 14. СИНДРОМ ПРАДЕРА-ВИЛЛИ у 70% больных наблюдается частичная делеция длинного плеча 15-й хромосомы (отцовская аллель), у 5%
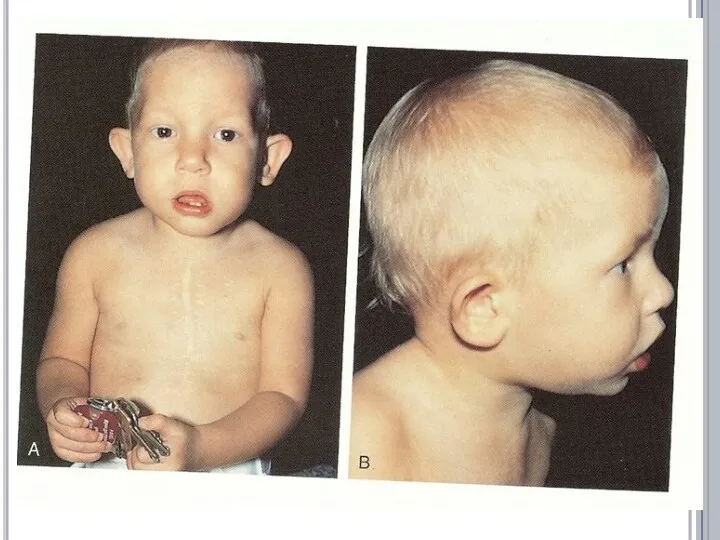
- 17. СИНДРОМ ДИ ДЖОРДЖИ Частичная моносомия 22q11.2. Популяционная частота - 1:20 000. Больные имеют следующие клинические признаки:
- 19. СИНДРОМ МАРТИНА - БЕЛЛА синдром ломкой, или фрагильной Х-хромосомы. Название синдрома объясняется особой формой строения Х-хромосомы,
- 20. Для больных характерны некоторые морфологические признаки, которые не всегда отчетливо проявляются (высокий выпуклый лоб, крупные уши
- 22. Скачать презентацию

СИНДРОМ ТРИСОМИИ ПО КОРОТКОМУ ПЛЕЧУ 9-Й ХРОМОСОМЫ
- наиболее частая форма
СИНДРОМ ТРИСОМИИ ПО КОРОТКОМУ ПЛЕЧУ 9-Й ХРОМОСОМЫ
- наиболее частая форма
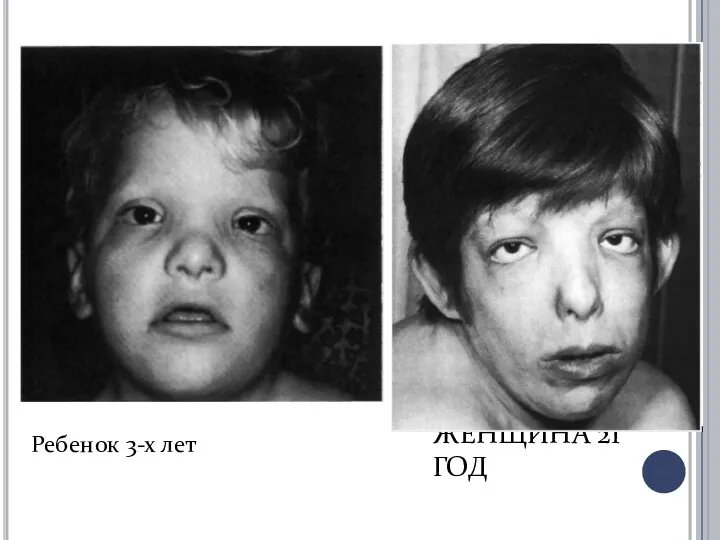
ЖЕНЩИНА 21 ГОД
Ребенок 3-х лет
ЖЕНЩИНА 21 ГОД
Ребенок 3-х лет

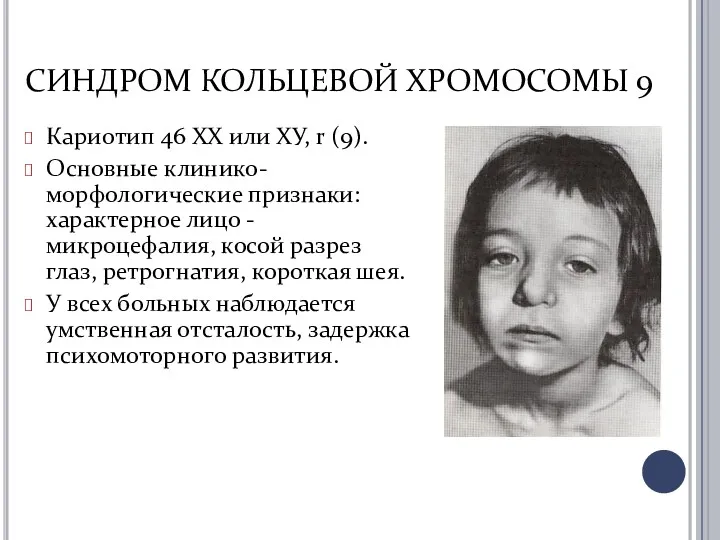
СИНДРОМ КОЛЬЦЕВОЙ ХРОМОСОМЫ 9
Кариотип 46 ХХ или ХУ, r (9).
Основные
СИНДРОМ КОЛЬЦЕВОЙ ХРОМОСОМЫ 9
Кариотип 46 ХХ или ХУ, r (9).
Основные

СИНДРОМ КОШАЧЬЕГО КРИКА
- частичная моносомия по короткому плечу хромосомы 5
СИНДРОМ КОШАЧЬЕГО КРИКА
- частичная моносомия по короткому плечу хромосомы 5

Клиническая картина синдрома 5р- довольно сильно различается у отдельных больных по
Клиническая картина синдрома 5р- довольно сильно различается у отдельных больных по
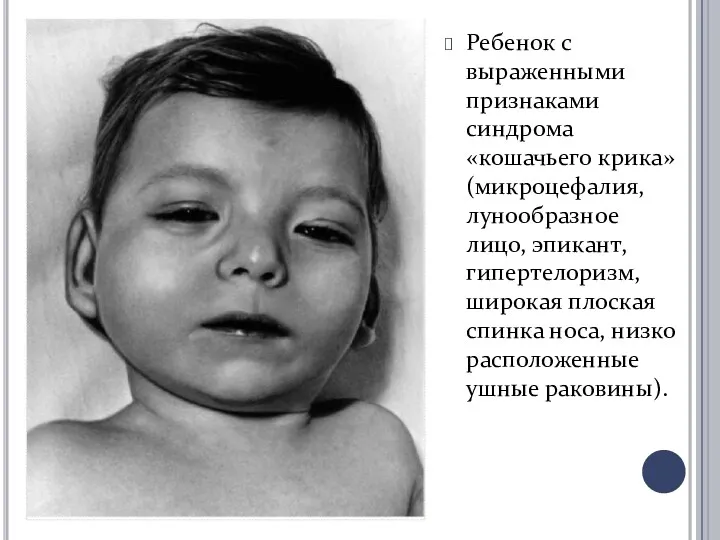
Ребенок с выраженными признаками синдрома «кошачьего крика» (микроцефалия, лунообразное лицо, эпикант,
Ребенок с выраженными признаками синдрома «кошачьего крика» (микроцефалия, лунообразное лицо, эпикант,
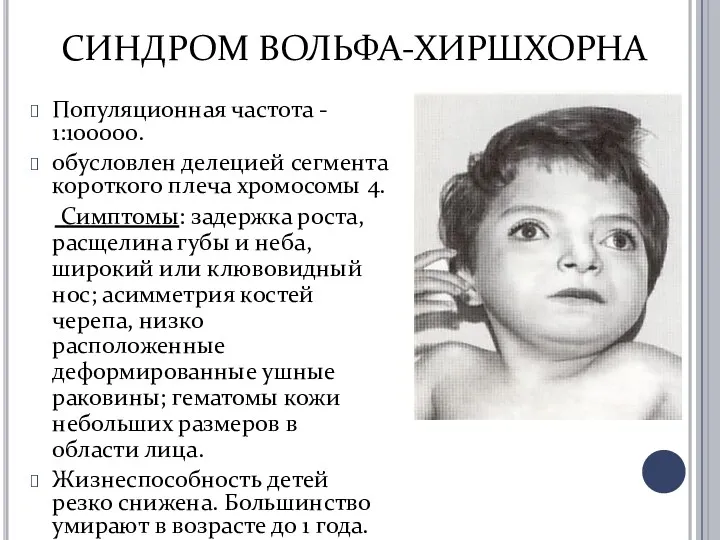
СИНДРОМ ВОЛЬФА-ХИРШХОРНА
Популяционная частота - 1:100000.
обусловлен делецией сегмента короткого плеча
СИНДРОМ ВОЛЬФА-ХИРШХОРНА
Популяционная частота - 1:100000.
обусловлен делецией сегмента короткого плеча


СИНДРОМ ОРБЕЛИ
обусловлен делецией длинного плеча 13-й хромосомы. Популяционная частота не
СИНДРОМ ОРБЕЛИ
обусловлен делецией длинного плеча 13-й хромосомы. Популяционная частота не
СИНДРОМ ПРАДЕРА-ВИЛЛИ
у 70% больных наблюдается частичная делеция длинного плеча 15-й
СИНДРОМ ПРАДЕРА-ВИЛЛИ
у 70% больных наблюдается частичная делеция длинного плеча 15-й

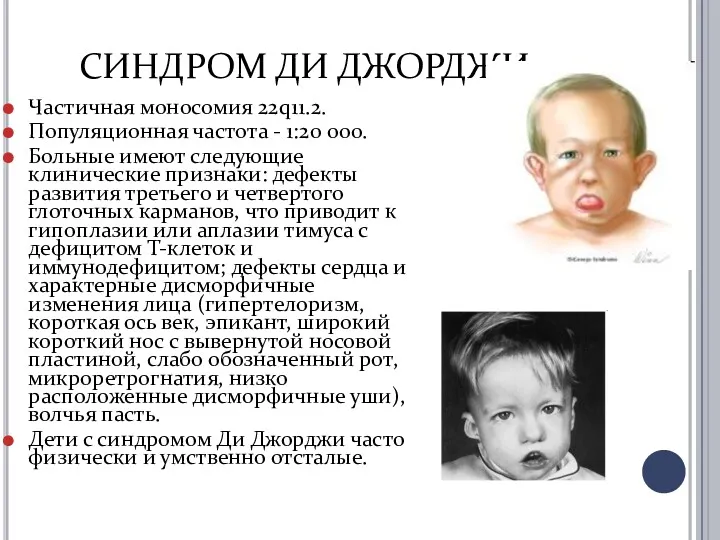
СИНДРОМ ДИ ДЖОРДЖИ
Частичная моносомия 22q11.2.
Популяционная частота - 1:20 000.
Больные имеют следующие
СИНДРОМ ДИ ДЖОРДЖИ
Частичная моносомия 22q11.2.
Популяционная частота - 1:20 000.
Больные имеют следующие
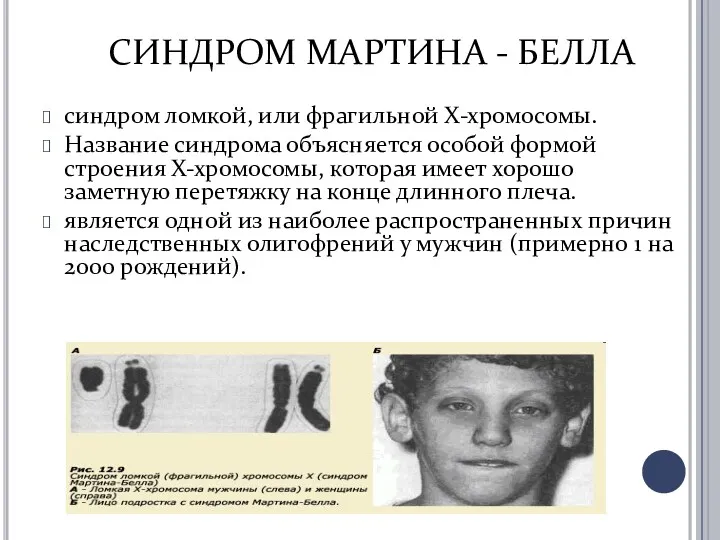
СИНДРОМ МАРТИНА - БЕЛЛА
синдром ломкой, или фрагильной Х-хромосомы.
Название синдрома объясняется особой
СИНДРОМ МАРТИНА - БЕЛЛА
синдром ломкой, или фрагильной Х-хромосомы.
Название синдрома объясняется особой

Для больных характерны некоторые морфологические признаки, которые не всегда отчетливо проявляются
Для больных характерны некоторые морфологические признаки, которые не всегда отчетливо проявляются
 Система комп’ютерного моделювання процесів життєдіяльності органів та систем організму СКІФ
Система комп’ютерного моделювання процесів життєдіяльності органів та систем організму СКІФ История болезни. Методы обследования больного
История болезни. Методы обследования больного Правила здорового образа жизни
Правила здорового образа жизни Подкомиссия врачебной комиссии по экспертизе профпригодности
Подкомиссия врачебной комиссии по экспертизе профпригодности Рак шейки матки. Основные ошибки гинекологов с позиции онколога. Органосохраняющие операции
Рак шейки матки. Основные ошибки гинекологов с позиции онколога. Органосохраняющие операции Қатты дәрілерді еріту, оларды флаконнан шприцке алу
Қатты дәрілерді еріту, оларды флаконнан шприцке алу ЛФК и массаж при ожирении
ЛФК и массаж при ожирении Лучевая болезнь животных
Лучевая болезнь животных Подготовка беременных к родам
Подготовка беременных к родам Операции на кровеносных сосудах
Операции на кровеносных сосудах Боткин Сергей Петрович
Боткин Сергей Петрович ДНҚ репликациясы
ДНҚ репликациясы Иммунный ответ. Определение, виды, особенности врожденного и адаптивного иммунитета
Иммунный ответ. Определение, виды, особенности врожденного и адаптивного иммунитета Профессиональная деятельность медсестры кабинета здорового ребенка
Профессиональная деятельность медсестры кабинета здорового ребенка Country slides measles
Country slides measles Средства, влияющие на адренергические синапсы
Средства, влияющие на адренергические синапсы Ожирение - заболевание или эстетическая проблема
Ожирение - заболевание или эстетическая проблема Болезни гипофиза: МСЭ и реабилитация
Болезни гипофиза: МСЭ и реабилитация Гинекологическое обследование детей и подростков в норме и при патологии
Гинекологическое обследование детей и подростков в норме и при патологии Стоматиты у детей
Стоматиты у детей Профессиограмма профессии Ветеринарный врач
Профессиограмма профессии Ветеринарный врач Туберкулез почек, мочевыводящей системы и мужских половых органов
Туберкулез почек, мочевыводящей системы и мужских половых органов Пути введения лекарственных средств
Пути введения лекарственных средств Развитие и функциональная анатомия половой системы
Развитие и функциональная анатомия половой системы Сестринский уход при болезнях пищеварения у гериатрических пациентов
Сестринский уход при болезнях пищеварения у гериатрических пациентов Лекарственные растения
Лекарственные растения Ауруханаға дейінгі кезеңдегі Анафилактикалық шоктың диагностикалық алгоритмі, шұғыл емі
Ауруханаға дейінгі кезеңдегі Анафилактикалық шоктың диагностикалық алгоритмі, шұғыл емі Болезнь Такаясу (неспецифический аортоартериит, синдром дуги аорты)
Болезнь Такаясу (неспецифический аортоартериит, синдром дуги аорты)