- Предмет и история геохимии

Содержание
- 2. Введение 1. Общие вопросы геохимии 2. Геохимия геологических процессов Геохимия эндогенных процессов Геохимия экзогенных процессов 3.
- 3. ГЕОХИМИЯ - наука о распространённости химических элементов и их изотопов в природе, процессах, определяющих формы их
- 4. Научные задачи геохимии: 1. Изучение форм нахождения элементов, их миграции и концентрации в геосферах Земли и
- 5. История геохимии Предистория (до появления в печати термина ―геохимия, Х.Ф. Шѐнбейн, 1838? 1842?) XVII век (18
- 6. История геохимии (после появления в печати термина геохимия, Х.Ф. Шенбейн, 1838? 1842?) XIX век (вторая половина,
- 7. Владимир Иванович Вернадский 1863-1945 Минералог и кристаллограф, основоположник геохимии, биогеохимии, радиогеологии и учения о биосфере, организатор
- 8. Предмет геохимии “В наших целях каждая порода может рассматриваться как химическая система, в которой под действием
- 9. История геохимии – ХХ век - Выдающиеся геохимики А.Е. Ферсман (1883–1945) – один из основателей современной
- 10. История геохимии – ХХ век – направления и лидеры В изучение химических процессов на Земле в
- 11. Некоторые значимые организации, издания и мероприятия мирового значения Первый выпуск журнала «Geochimica et Cosmochimica Acta» появился
- 12. Основные разделы геохимии (самостоятельные дисциплины) 1. Общая геохимия и космохимия 2. Геохимия изотопов 3.Физическая геохимия 4.
- 13. Повсеместное распространение ХЭ во всех геосферах Непрерывная миграция ХЭ во времени и в пространстве Многообразие форм
- 14. Хронология открытия химических элементов до 1500 г. (13 элементов): Cu, Ag, Au, Pb, Sn, Fe, C
- 15. Хронология открытия химических элементов
- 16. Периодическая система (классический вид)
- 17. Периодическая система (современный вид) A = Z + N называется массовым числом. Ядра с одинаковым Z,
- 18. Периодическая система (современный вид)
- 19. В настоящее время периодическая система охватывает 126 элементов. Из них все трансурановые элементы (Z = 93-107),
- 20. «Средние содержания ряда ведущих элементов в земной коре исследовались с 1815 английским учёным У. Филлипсом. Обобщение
- 21. ПРАВИЛА РАСПРОСТРАНЕННОСТИ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ 1. Все доступное для исследования вещество состоит из одних и тех же
- 22. Распространенность химических элементов в Солнечной системе
- 23. 3. Космическая распространенность химических элементов определяется стабильностью ядер атомов (Вернадский, 1921, Goldschmidt, 1930). Среди наиболее существенных
- 24. Распространенность элементов в фотосфере Солнца ( Anders , Grevesse , 1989)
- 25. 4. Химические элементы образуются в ходе ядерных процессов (процессов нуклеосинтеза), протекающих на разных стадиях эволюции Вселенной
- 26. Основные этапы эволюции массивной звезды (М > 25 М○)
- 27. Теоретический расчет возможных ядерных реакций в звездах различной массы Возможные ядерные реакции Нет Горение водорода Горение
- 28. Есть предположения, что синтез химических элементов непрерывно идет во Вселенной. На Земле новые элементы, возможно, образуются
- 29. ПРОЦЕССЫ НУКЛЕОСИНТЕЗА Источником большинства ядер является определенная последовательность ядерных реакций, протекающих в недрах звезд.
- 30. РЕАКЦИИ НУКЛЕОСИНТЕЗА Горение водорода. Это один из основных процессов, поддерживающих длительное выделение энергии в звездах. При
- 32. Горение гелия. После того, как в звезде накапливается гелий, под действием сил гравитации гелиевое ядро сжимается,
- 33. α-Процесс. Это процесс последовательного добавления α-частиц к ядру 20Ne с образованием ядер 24Mg, 28Si, 32S, 36Ar,
- 34. E-процесс. Во взрывном Нуклеосинтезе сеть ядерных реакций (рис. 1), протекающих при Т ~ 3 х 109
- 35. НУКЛЕОСИНТЕЗ В СВЕРХНОВЫХ – ВОЗНИКНОВЕНИЕ ПИКА ЖЕЛЕЗА НА КОНЕЧНОЙ СТАДИИ ЭВОЛЮЦИИ ЗВЕЗД Сверхновые звезды - это
- 36. Эта стадия в эволюции массивной звезды наступает тогда, когда завершаются реакции термоядерного синтеза и в центре
- 37. При взрывном горении кремния в оболочке сверхновой вне коллапсирующего ядра, но в непосредственной близости от него
- 38. Наблюдения указывают на спад светимости после максимума блеска с характерным временем, близким ко времени распада 56Ni
- 39. Другие процессы s-Процесс. Это образование ядер тяжелее железа в результате медленного последовательного захвата нейтронов. Скорость s-процесса
- 40. Лекция 3. Космохимические основания геохимии. Классификация метеоритов 1. Самым распространенным типом метеоритов являются хондриты, которые отличаются
- 43. Углистый хондрит Обыкновенный хондрит Ахондрит Железный метеорит
- 44. Распространенность химических элементов (число атомов ni на 106 атомов Si) в CI-хондритах.
- 45. Соотношения распространенности химических элементов (число атомов ni на 106 атомов Si) на Солнце (ns) и в
- 46. Лекция 3. Космохимические основания геохимии 3. Формирование твердых фаз протопланетного вещества Солнечной системы сопровождалось фракционированием химических
- 47. Геохимическая классификация элементов В.М. Гольдшмидта (Goldschmidt, 1924)
- 48. Лекция 3. Космохимические основания геохимии 4. Итогом космохимической эволюции протопланетного вещества является формирование четырех типов фаз
- 49. Твердые фазы протопланетного вещества
- 50. Лекция 3. Космохимические основания геохимии 5. Классические данные по изотопному составу Pb и новые данные по
- 51. Лекция 3. Космохимические основания геохимии 7. Главными продуктами планетной дифференциации рассматриваются базальты кор планет земной группы;
- 52. Геохимические особенности базальтов Земли, Луны и родительских тел шерготтитов (Марс?) и эвкритов
- 53. Лекция 4. Строение и химический состав оболочек Земли. Под моделью внутреннего строения Земли понимают разрез планеты,
- 54. Распределение скоростей сейсмических волн (Vp, Vs) с глубиной согласно стандартной геофизической модели Земли
- 55. Схема глубинного строения Земли (по К.Е.Буллену) В начале 40-ых годов прошлого столетия австралийский сейсмолог К.Е.Буллен (Keith
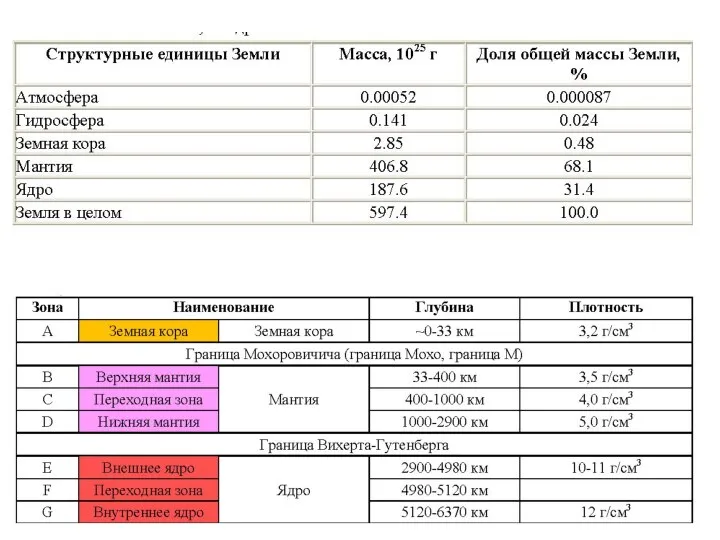
- 56. Распределение давления (Р) и плотности (?) с глубиной согласно стандартной геофизической модели Земли Значение средней плотности
- 58. Строение земной коры
- 59. 1. Экстраполяция плотности глубинного вещества к нормальному давлению показывает, что она (кроме вещества верхней мантии) не
- 60. Состав вещества внутренних оболочек Земли 3. Альтернатива - формирование Земли в результате аккреции недифференцированной примитивной твердой
- 61. 4. Экспериментальные данные о плотности Fe, Ni-сплава при давлениях, отвечающих земному ядру, показывают, что для корректного
- 62. Химический состав ядра Химический состав мантии Земли
- 63. Лерцолит Дунит
- 64. Полиморфизм Впервые идея о возможности оливина под действием высоких давлений принимать структуру шпинели и увеличивать тем
- 65. Полиморфизм важнейших фаз земного вещества
- 66. Шпинель Гранат Ильменит Перовскит
- 67. 6. Распространенность химических элементов в верхней мантии Земли, оцениваемая на основании данных о их распространенности в
- 68. Распространенность химических элементов (ni/106 Si) в H-, L-, LL-хондритах (Wasson, Kallemeyn, 1988) и примитивной мантии Земли
- 69. Состав вещества внутренних оболочек Земли 7. Изотопные данные заставляют предполагать, что геохимическая гетерогенность верхней мантии (силикатной
- 70. Распределение химических элементов в системе хондриты (Х) - дуниты мантии (УО) - базальты (Б) и граниты
- 71. Из рассмотренного материала можно прийти к следующим заключениям: В состав Земли при ее формировании в ходе
- 72. Основной химический состав геосфер
- 73. Порядки содержаний ХЭ в земной коре (г/т)
- 74. Лекция 5. Строение и химический состав земной коры
- 75. Скорость распространения сейсмических волн в континентальной земной коре и наиболее распространенных горных породах.
- 76. Распространенность горных пород осадочной оболочки континентов и океанов и гранитно-метаморфической оболочки континентов.
- 77. Земная кора 1. Средний химический состав земной коры отвечает средневзвешенному составу продуктов выплавления и дегазации мантии
- 78. Распределение содержаний SiO2 в магматических породах (Richardson, Sneesby, 1923)
- 79. 4. Статистика составов магматических пород позволила оценить относительную распространенность различных их типов в верхней части континентальной
- 80. Земная кора 6. Альтернативные модели химического состава земной коры основаны на данных о реальной распространенности главнейших
- 81. Земная кора 7. Состав гранулит-базитового слоя континентальной коры должен отличаться от состава гранитно-метаморфического слоя, по-видимому, более
- 82. Земная кора 8. Оценка химического состава континентальной коры в целом остается неопределенной из-за незнания состава гранулитбазитового
- 83. Распространенность химических элементов в верхней части континентальной коры, 10-4 вес. %
- 84. Земная кора 9. Средний химический состав гранитно-метаморфического слоя (верхней части континентальной коры) отличается от модельного состава
- 85. Земная кора Континентальная кора существенно обогащена SiO2 и литофильными элементами с большим ионным радиусом, напр. U,
- 86. Земная кора Выведенные на поверхность осадочные и магматич. породы подвергаются выветриванию – воздействию разрушающих агентов, в
- 87. Лекция 6. Многообразие форм и видов нахождения химических элементов в природе Определяется минеральной формой существования элементов
- 88. Минеральная форма. В земной коре установлено более 4000 минеральных форм, что значительно меньше теоретически возможных. Причиной
- 89. Корреляция числа минералов (Ni) химических элементов и их распространенности в верхней части континентальной коры (ni)
- 90. Способность химических элементов к минералообразованию
- 91. Типы и классы минералов по химическому составу (в скобках — приблизительное количество минералов
- 92. Формы нахождения химических элементов в рудах Для основных рудных элементов характерна минеральная форма. Для элементов-примесей с
- 93. Участок Нембондачан (Au-Ag-полиметаллические руды) Участок Нембондачан (Ag-полиметаллические руды) Участок Нембондачан (полиметаллическая ассоциация) Медные руды Свинцовые руды
- 94. Т.н. 58/19 – образец жильного кварца с арсенопиритом и самородным золотом (Au=52 г/т) Т.н. 58/32– образец
- 95. Агрегат лёллингита, арсенопирита, борнита, халькопирита и теннантита-тетраэдрита, обр. 72-31/5. Участок Рыжий А) Борнит с решетчатыми структурами
- 96. Самородное золото участка Туманный золота Теллуриды золота и серебра участка Вукней
- 97. Образцы руд с сульфидно-сульфоантимонидной минерализацией с участка Вернитакайвеем Оруденелые метасоматиты Кварцевые прожилки в оруденелых метасоматитах
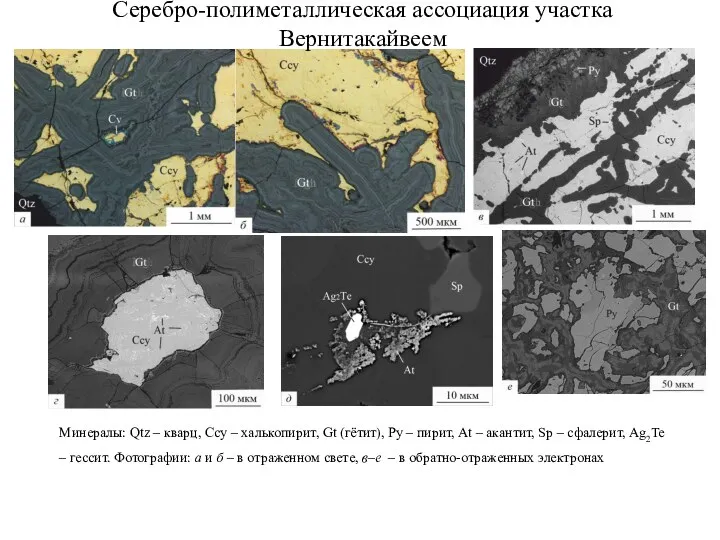
- 98. Серебро-полиметаллическая ассоциация участка Вернитакайвеем
- 99. Золото-серебряная ассоциация участка Вернитакайвеем Россыпное золото 794%o Plg Плагионит – Pb5Sb8S17 Zk Цинкенит – Pb6Sb14S27 Tt
- 100. а) Вростки золота в пирите, б) электрум (пробность 684) в интерстициях между зернами пирита, в) вростки
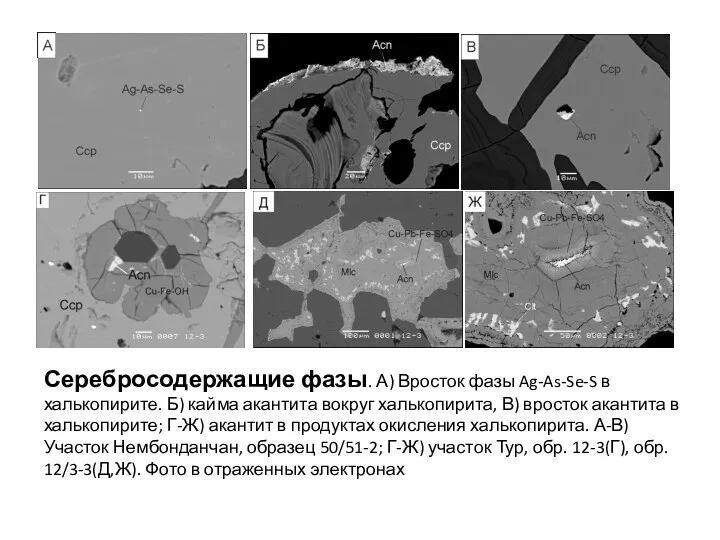
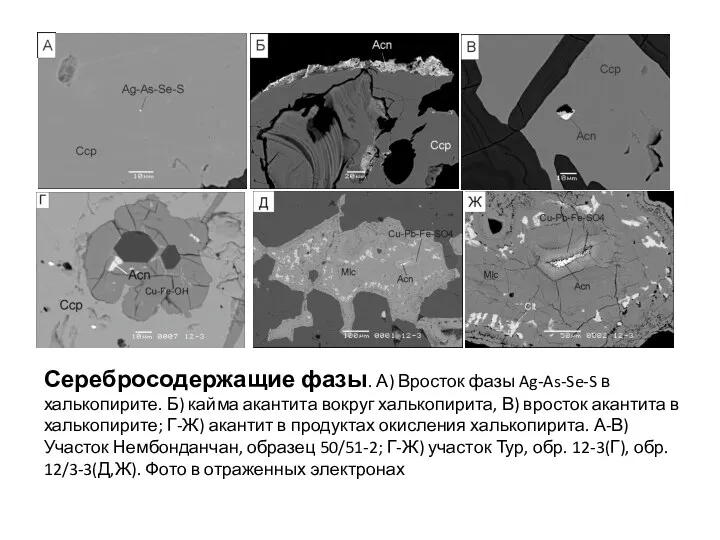
- 101. Ри Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма акантита вокруг халькопирита, В) вросток
- 102. Ри Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма акантита вокруг халькопирита, В) вросток
- 103. Минералы карбонат-кварцевых жил. А, Б, Г) Гнезда карбонатов марганца (замещены оксидами и гидроксидами марганца сложного состава)
- 104. Геохимические запасы важнейших металлов в континентальной коре до глубины 1 км и в промышленных месторождениях
- 105. Корреляционная зависимость между кларками земной коры и запасами элементов в месторождениях одинаковой крупности
- 106. Минеральная и безминеральная формы нахождения элементов в литосфере, %
- 107. Безминеральная форма. Наиболее характерными безминеральными формами нахождения элементов в природе являются следующие: природные истинные и коллоидные
- 108. Лекция 7. Всеобщая миграция химических элементов во времени и пространстве Космическим рассеянием; Перегруппировкой химических элементов между
- 109. Миграция химических элементов Находит отражение в гигантских тектоно-магматических процессах, преобразующих земную кору, и в тончайших химических
- 110. Схема геологического цикла миграции химических элементов
- 111. Существует два механизма переноса химических элементов в геологических процессах - молекулярная диффузия и конвективное перераспределение фаз
- 112. Конвективный массоперенос Конвекция – физико-химическая миграция атомов, ионов, молекул вместе с растворителем. Конвекция в пористой среде
- 113. Диффузионный массоперенос Диффузия – физико-химическая миграция вещества для установления равновесных концентраций вследствие беспорядочного движения атомов, ионов,
- 114. Гипогенная зона характеризуется высокими и сверхвысокими температурами, давлением и концентрацией химических элементов, что приводит к метаморфизации
- 115. Гипергенная зона Зона гипергенеза является главным местом действия солнечной радиации. Под ее влиянием прямо или косвенно
- 116. В магматических процессах основной вклад в перераспре-деление химических элементов вносит разделение твердых и жидкой фаз, т.
- 118. Скачать презентацию

Введение
1. Общие вопросы геохимии
2. Геохимия геологических процессов
Геохимия эндогенных процессов
Геохимия экзогенных процессов
3.
Введение
1. Общие вопросы геохимии
2. Геохимия геологических процессов
Геохимия эндогенных процессов
Геохимия экзогенных процессов
3.

ГЕОХИМИЯ - наука о распространённости химических элементов и их изотопов в природе,
ГЕОХИМИЯ - наука о распространённости химических элементов и их изотопов в природе,

Научные задачи геохимии:
1. Изучение форм нахождения элементов, их миграции и концентрации
Научные задачи геохимии:
1. Изучение форм нахождения элементов, их миграции и концентрации

История геохимии
Предистория (до появления в печати термина ―геохимия, Х.Ф. Шѐнбейн, 1838?
История геохимии
Предистория (до появления в печати термина ―геохимия, Х.Ф. Шѐнбейн, 1838?

История геохимии (после появления в печати термина геохимия, Х.Ф. Шенбейн, 1838?
История геохимии (после появления в печати термина геохимия, Х.Ф. Шенбейн, 1838?

Владимир Иванович Вернадский
1863-1945
Минералог и кристаллограф, основоположник геохимии, биогеохимии, радиогеологии и учения
Владимир Иванович Вернадский
1863-1945
Минералог и кристаллограф, основоположник геохимии, биогеохимии, радиогеологии и учения

Предмет геохимии
“В наших целях каждая порода может рассматриваться как химическая система,
Предмет геохимии
“В наших целях каждая порода может рассматриваться как химическая система,

История геохимии – ХХ век - Выдающиеся геохимики
А.Е. Ферсман (1883–1945)
История геохимии – ХХ век - Выдающиеся геохимики
А.Е. Ферсман (1883–1945)

История геохимии – ХХ век – направления и лидеры
В изучение химических
История геохимии – ХХ век – направления и лидеры
В изучение химических

Некоторые значимые организации, издания и мероприятия мирового значения
Первый выпуск журнала «Geochimica
Некоторые значимые организации, издания и мероприятия мирового значения
Первый выпуск журнала «Geochimica

Основные разделы геохимии (самостоятельные дисциплины)
1. Общая геохимия и космохимия
2. Геохимия изотопов
3.Физическая
Основные разделы геохимии (самостоятельные дисциплины)
1. Общая геохимия и космохимия
2. Геохимия изотопов
3.Физическая
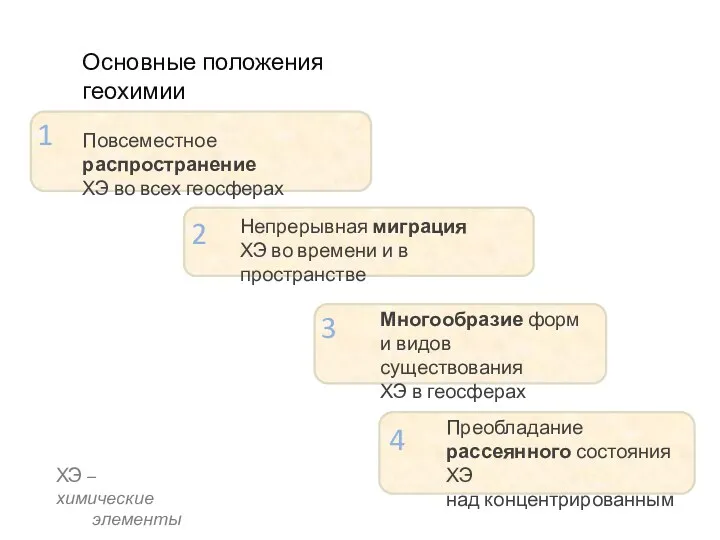
Повсеместное распространение
ХЭ во всех геосферах
Непрерывная миграция
ХЭ во времени и
Повсеместное распространение
ХЭ во всех геосферах
Непрерывная миграция
ХЭ во времени и

Хронология открытия химических элементов
до 1500 г. (13 элементов): Cu, Ag, Au, Pb,
Хронология открытия химических элементов
до 1500 г. (13 элементов): Cu, Ag, Au, Pb,
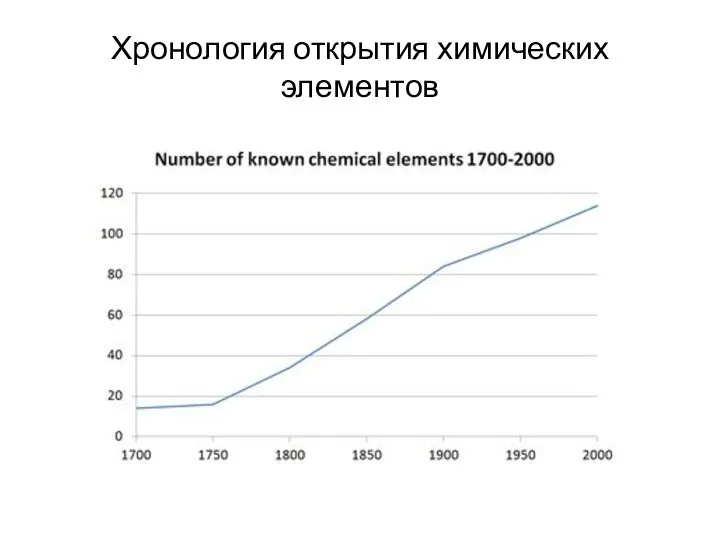
Хронология открытия химических элементов
Хронология открытия химических элементов
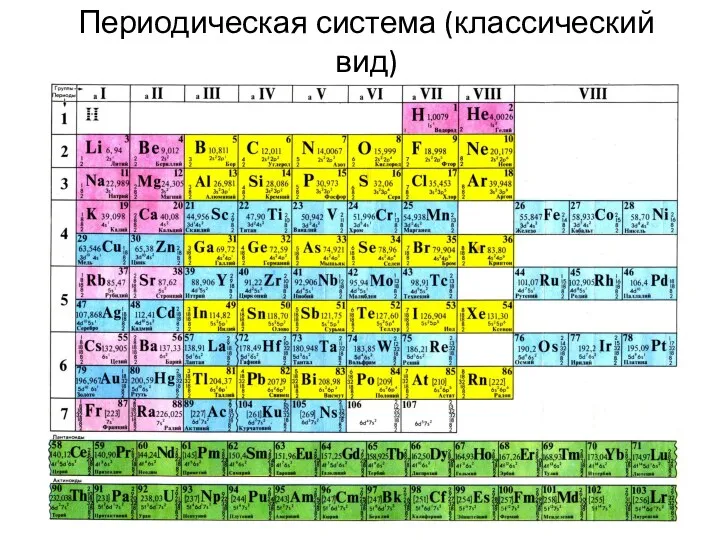
Периодическая система (классический вид)
Периодическая система (классический вид)
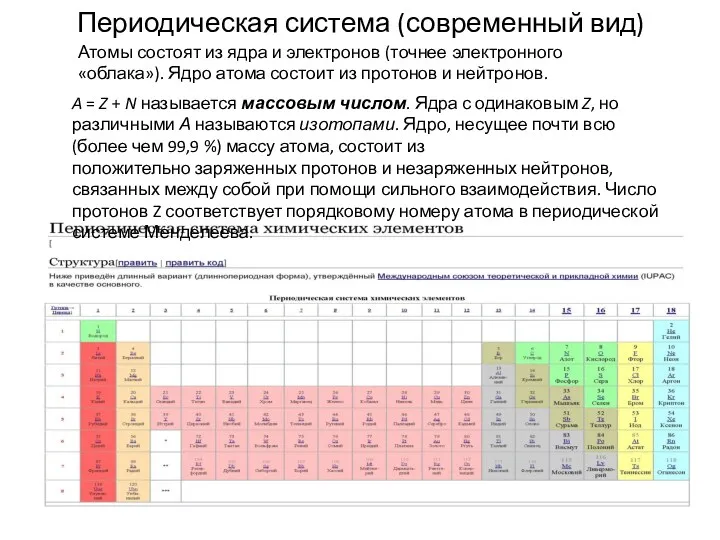
Периодическая система (современный вид)
A = Z + N называется массовым числом. Ядра с одинаковым Z, но различными А называются изотопами. Ядро, несущее
Периодическая система (современный вид)
A = Z + N называется массовым числом. Ядра с одинаковым Z, но различными А называются изотопами. Ядро, несущее

Периодическая система (современный вид)
Периодическая система (современный вид)

В настоящее время периодическая система охватывает 126 элементов. Из них все
В настоящее время периодическая система охватывает 126 элементов. Из них все

«Средние содержания ряда ведущих элементов в земной коре исследовались с
«Средние содержания ряда ведущих элементов в земной коре исследовались с

ПРАВИЛА РАСПРОСТРАНЕННОСТИ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
1. Все доступное для исследования вещество состоит из
ПРАВИЛА РАСПРОСТРАНЕННОСТИ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
1. Все доступное для исследования вещество состоит из
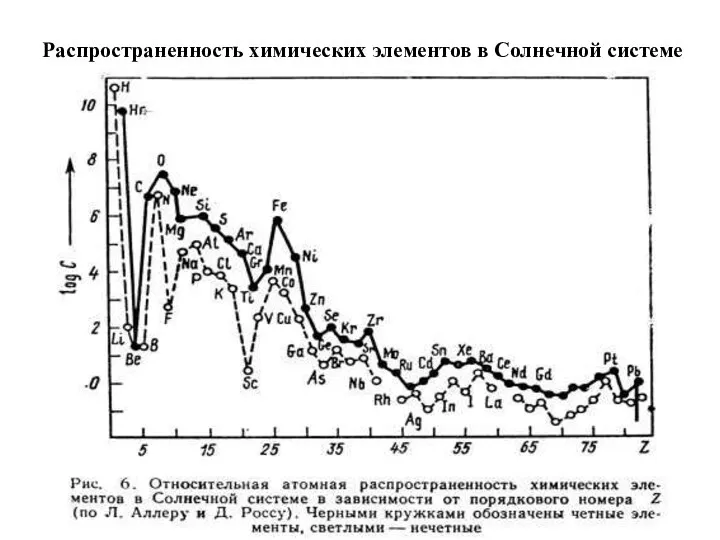
Распространенность химических элементов в Солнечной системе
Распространенность химических элементов в Солнечной системе

3. Космическая распространенность химических элементов определяется стабильностью ядер атомов (Вернадский, 1921,
3. Космическая распространенность химических элементов определяется стабильностью ядер атомов (Вернадский, 1921,
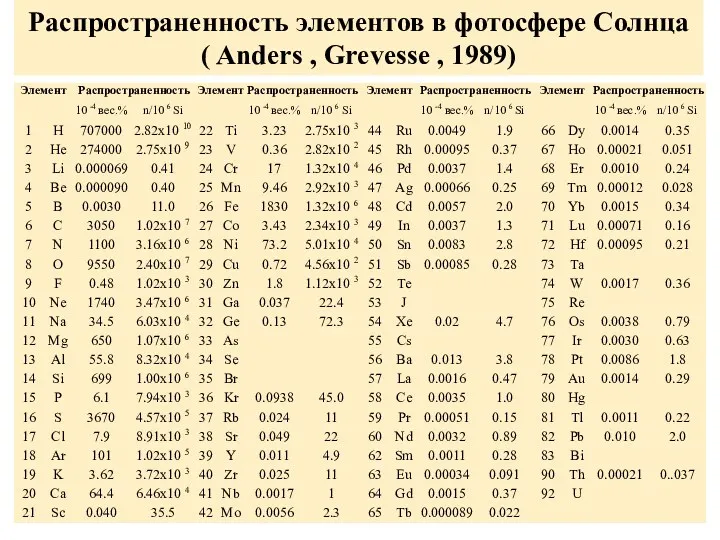
Распространенность элементов в фотосфере Солнца
( Anders , Grevesse , 1989)
Распространенность элементов в фотосфере Солнца
( Anders , Grevesse , 1989)

4. Химические элементы образуются в ходе ядерных процессов (процессов нуклеосинтеза), протекающих
4. Химические элементы образуются в ходе ядерных процессов (процессов нуклеосинтеза), протекающих
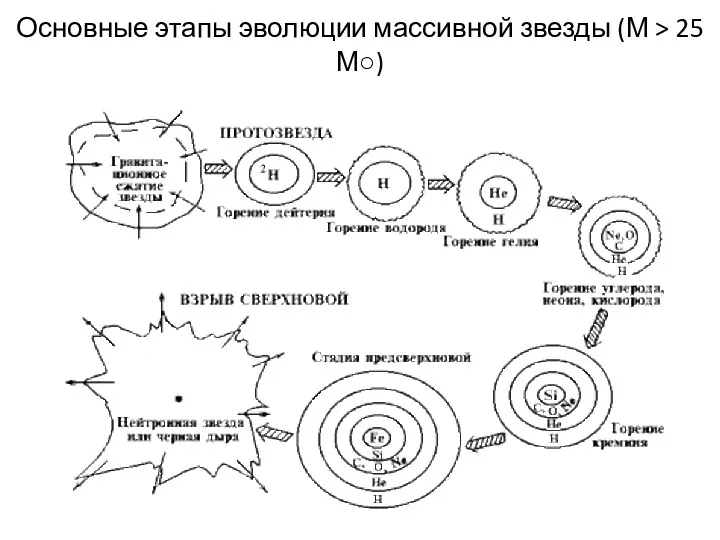
Основные этапы эволюции массивной звезды (М > 25 М○)
Основные этапы эволюции массивной звезды (М > 25 М○)

Теоретический расчет возможных ядерных реакций в звездах различной массы
Возможные ядерные реакции
Нет
Горение
Теоретический расчет возможных ядерных реакций в звездах различной массы
Возможные ядерные реакции
Нет
Горение

Есть предположения, что синтез химических элементов непрерывно идет во Вселенной. На
Есть предположения, что синтез химических элементов непрерывно идет во Вселенной. На
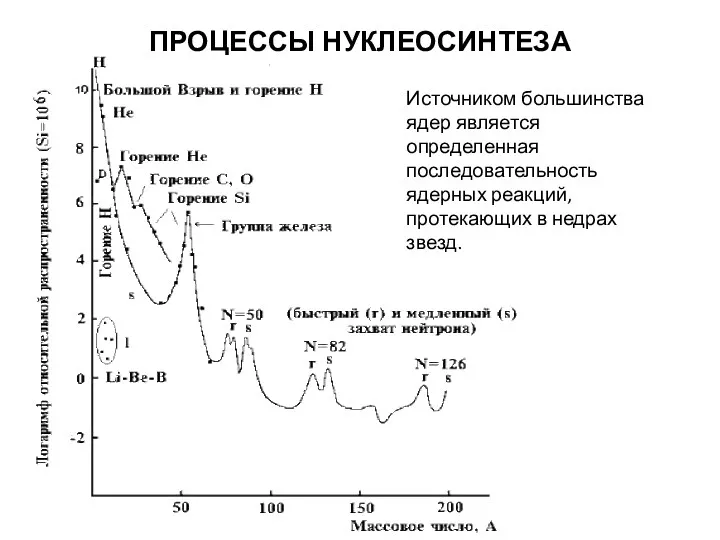
ПРОЦЕССЫ НУКЛЕОСИНТЕЗА
Источником большинства ядер является определенная последовательность ядерных реакций, протекающих в
ПРОЦЕССЫ НУКЛЕОСИНТЕЗА
Источником большинства ядер является определенная последовательность ядерных реакций, протекающих в

РЕАКЦИИ НУКЛЕОСИНТЕЗА
Горение водорода. Это один из основных процессов, поддерживающих длительное выделение
РЕАКЦИИ НУКЛЕОСИНТЕЗА
Горение водорода. Это один из основных процессов, поддерживающих длительное выделение
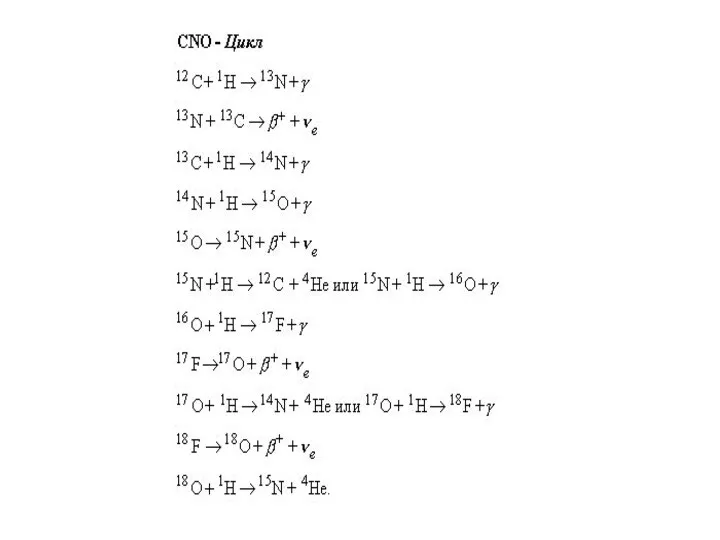

Горение гелия.
После того, как в звезде накапливается гелий, под действием сил
Горение гелия.
После того, как в звезде накапливается гелий, под действием сил

α-Процесс.
Это процесс последовательного добавления α-частиц к ядру 20Ne с образованием ядер 24Mg, 28Si, 32S, 36Ar, 40Ca. Он описывает
α-Процесс.
Это процесс последовательного добавления α-частиц к ядру 20Ne с образованием ядер 24Mg, 28Si, 32S, 36Ar, 40Ca. Он описывает
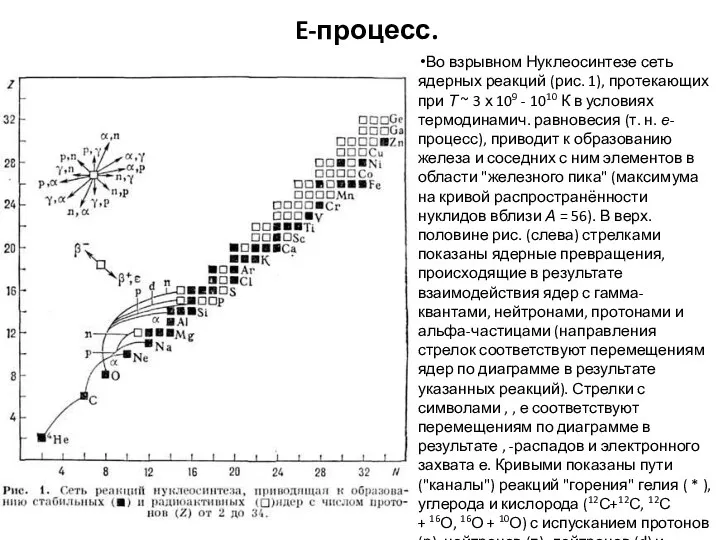
E-процесс.
Во взрывном Нуклеосинтезе сеть ядерных реакций (рис. 1), протекающих при Т ~
E-процесс.
Во взрывном Нуклеосинтезе сеть ядерных реакций (рис. 1), протекающих при Т ~
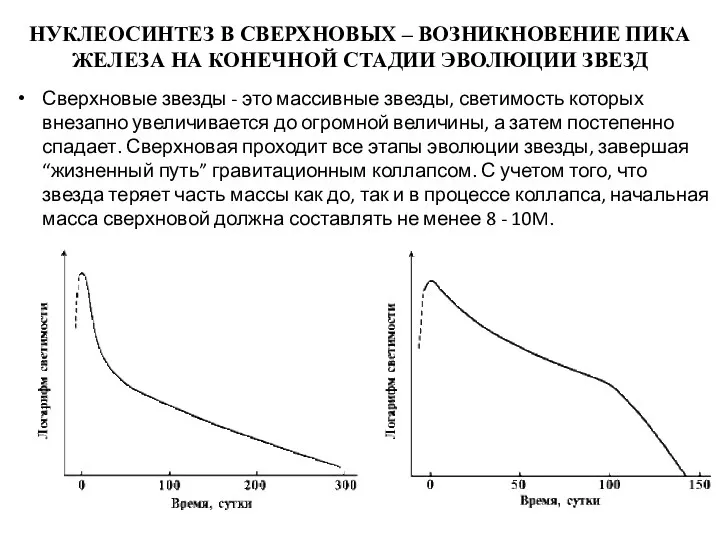
НУКЛЕОСИНТЕЗ В СВЕРХНОВЫХ – ВОЗНИКНОВЕНИЕ ПИКА ЖЕЛЕЗА НА КОНЕЧНОЙ СТАДИИ ЭВОЛЮЦИИ
НУКЛЕОСИНТЕЗ В СВЕРХНОВЫХ – ВОЗНИКНОВЕНИЕ ПИКА ЖЕЛЕЗА НА КОНЕЧНОЙ СТАДИИ ЭВОЛЮЦИИ

Эта стадия в эволюции массивной звезды наступает тогда, когда завершаются реакции
Эта стадия в эволюции массивной звезды наступает тогда, когда завершаются реакции

При взрывном горении кремния в оболочке сверхновой вне коллапсирующего ядра, но
При взрывном горении кремния в оболочке сверхновой вне коллапсирующего ядра, но

Наблюдения указывают на спад светимости после максимума блеска с характерным временем,
Наблюдения указывают на спад светимости после максимума блеска с характерным временем,

Другие процессы
s-Процесс. Это образование ядер тяжелее железа в результате медленного последовательного
Другие процессы
s-Процесс. Это образование ядер тяжелее железа в результате медленного последовательного

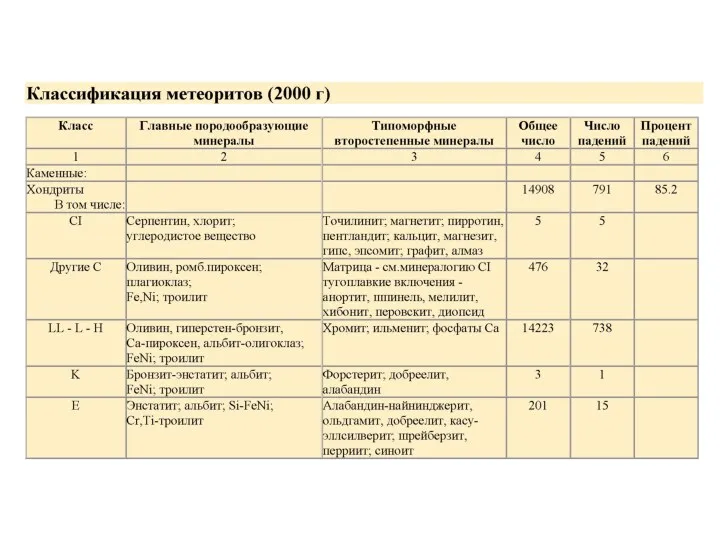
Лекция 3. Космохимические основания геохимии. Классификация метеоритов
1. Самым распространенным типом метеоритов
Лекция 3. Космохимические основания геохимии. Классификация метеоритов
1. Самым распространенным типом метеоритов
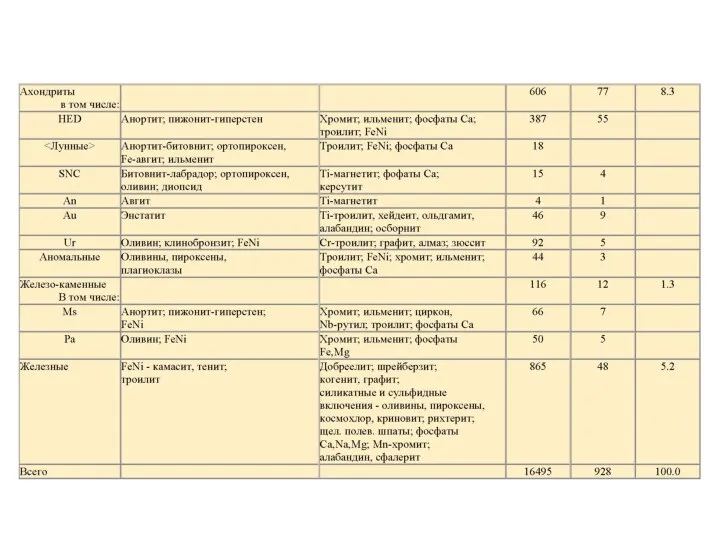

Углистый хондрит
Обыкновенный хондрит
Ахондрит
Железный метеорит
Углистый хондрит
Обыкновенный хондрит
Ахондрит
Железный метеорит
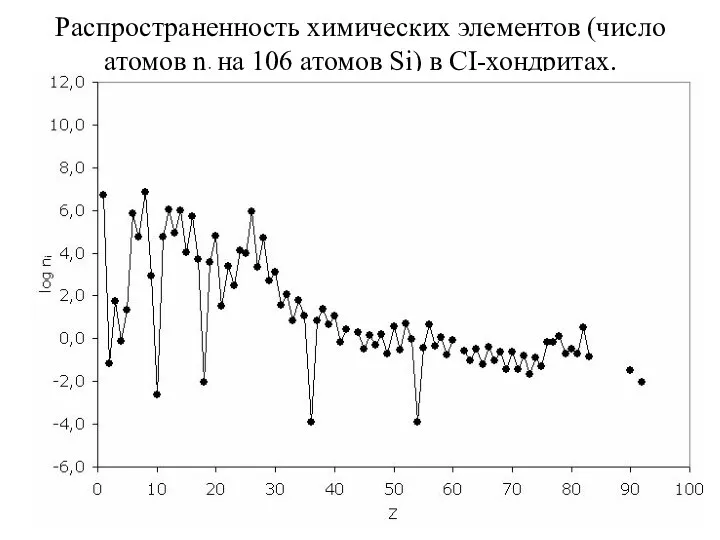
Распространенность химических элементов (число атомов ni на 106 атомов Si) в
Распространенность химических элементов (число атомов ni на 106 атомов Si) в
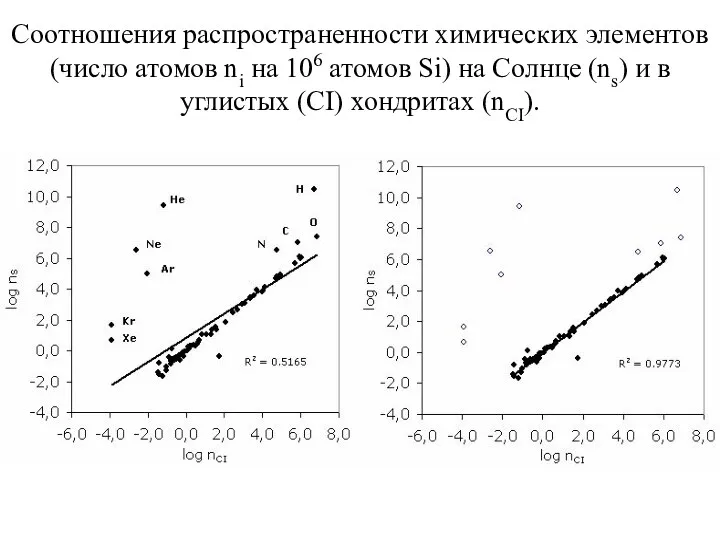
Соотношения распространенности химических элементов (число атомов ni на 106 атомов Si)
Соотношения распространенности химических элементов (число атомов ni на 106 атомов Si)

Лекция 3. Космохимические основания геохимии
3. Формирование твердых фаз протопланетного вещества Солнечной
Лекция 3. Космохимические основания геохимии
3. Формирование твердых фаз протопланетного вещества Солнечной

Геохимическая классификация элементов
В.М. Гольдшмидта (Goldschmidt, 1924)
Геохимическая классификация элементов
В.М. Гольдшмидта (Goldschmidt, 1924)

Лекция 3. Космохимические основания геохимии
4. Итогом космохимической эволюции протопланетного вещества является
Лекция 3. Космохимические основания геохимии
4. Итогом космохимической эволюции протопланетного вещества является
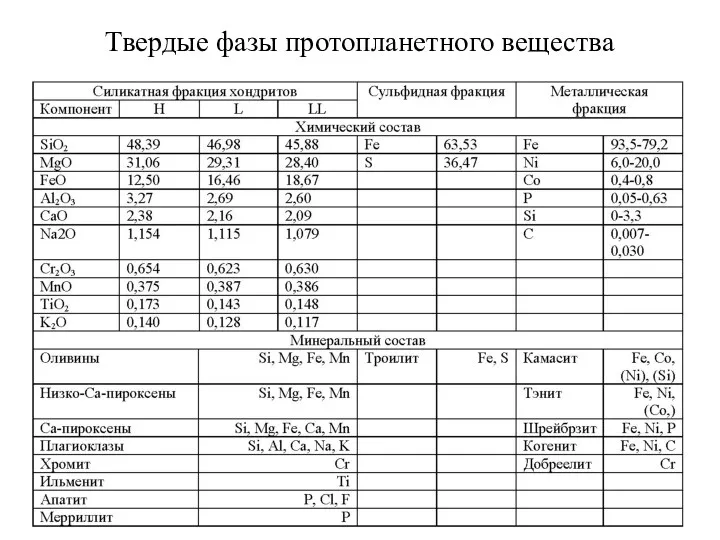
Твердые фазы протопланетного вещества
Твердые фазы протопланетного вещества

Лекция 3. Космохимические основания геохимии
5. Классические данные по изотопному составу Pb
Лекция 3. Космохимические основания геохимии
5. Классические данные по изотопному составу Pb

Лекция 3. Космохимические основания геохимии
7. Главными продуктами планетной дифференциации рассматриваются базальты
Лекция 3. Космохимические основания геохимии
7. Главными продуктами планетной дифференциации рассматриваются базальты
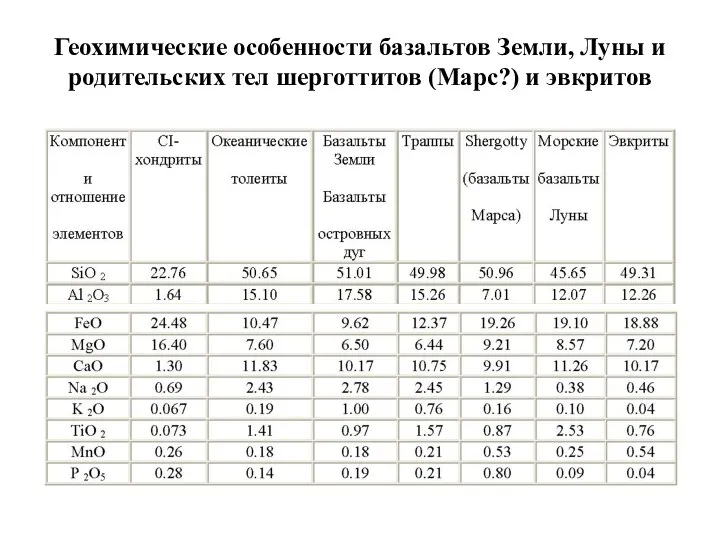
Геохимические особенности базальтов Земли, Луны и родительских тел шерготтитов (Марс?) и
Геохимические особенности базальтов Земли, Луны и родительских тел шерготтитов (Марс?) и
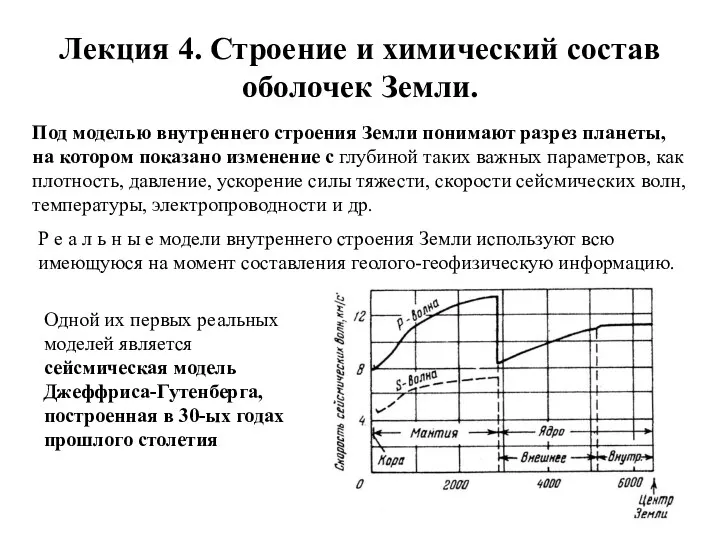
Лекция 4. Строение и химический состав оболочек Земли.
Под моделью внутреннего
Лекция 4. Строение и химический состав оболочек Земли.
Под моделью внутреннего
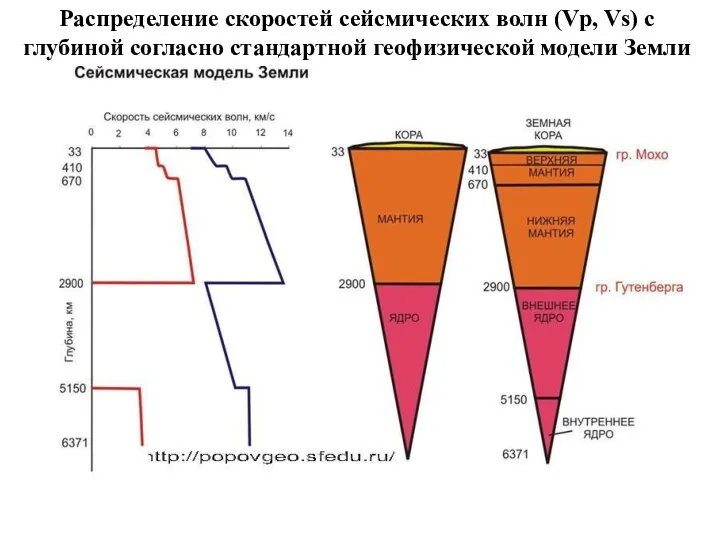
Распределение скоростей сейсмических волн (Vp, Vs) с глубиной согласно стандартной геофизической
Распределение скоростей сейсмических волн (Vp, Vs) с глубиной согласно стандартной геофизической
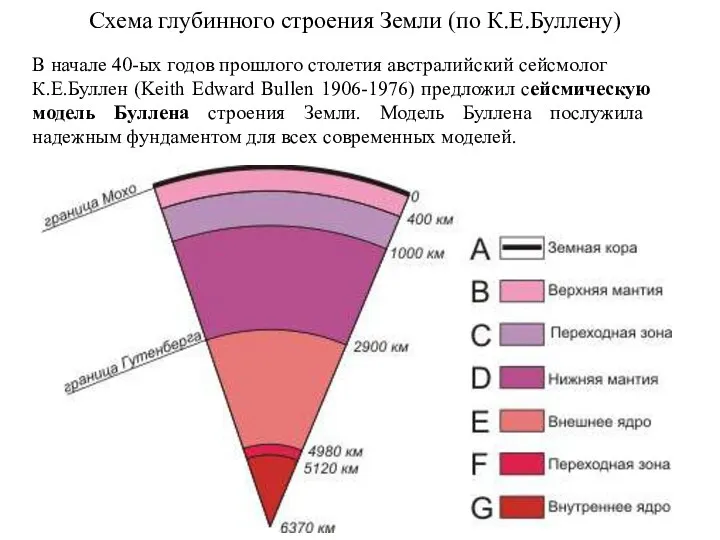
Схема глубинного строения Земли (по К.Е.Буллену)
В начале 40-ых годов прошлого столетия
Схема глубинного строения Земли (по К.Е.Буллену)
В начале 40-ых годов прошлого столетия
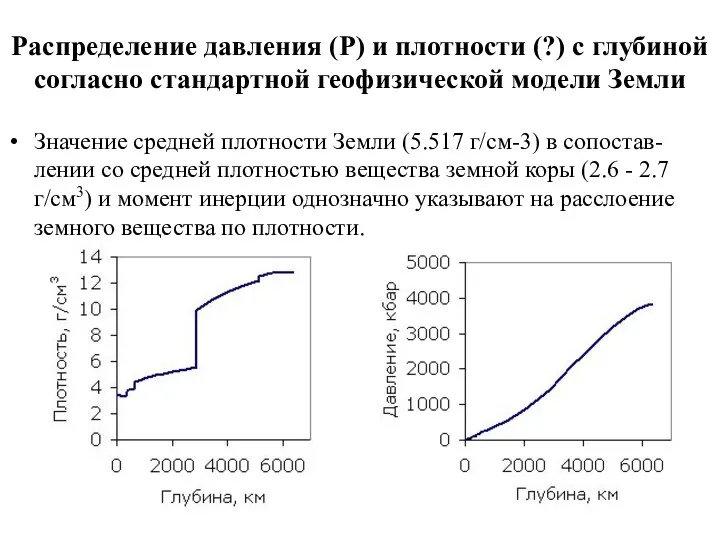
Распределение давления (Р) и плотности (?) с глубиной согласно стандартной геофизической
Распределение давления (Р) и плотности (?) с глубиной согласно стандартной геофизической
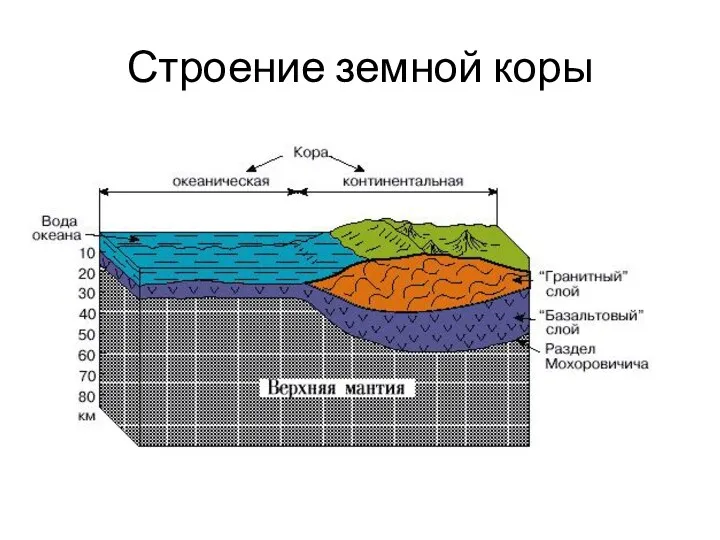
Строение земной коры
Строение земной коры

1. Экстраполяция плотности глубинного вещества к нормальному давлению показывает, что
1. Экстраполяция плотности глубинного вещества к нормальному давлению показывает, что

Состав вещества внутренних оболочек Земли
3. Альтернатива - формирование Земли
Состав вещества внутренних оболочек Земли
3. Альтернатива - формирование Земли

4. Экспериментальные данные о плотности Fe, Ni-сплава при давлениях, отвечающих земному
4. Экспериментальные данные о плотности Fe, Ni-сплава при давлениях, отвечающих земному
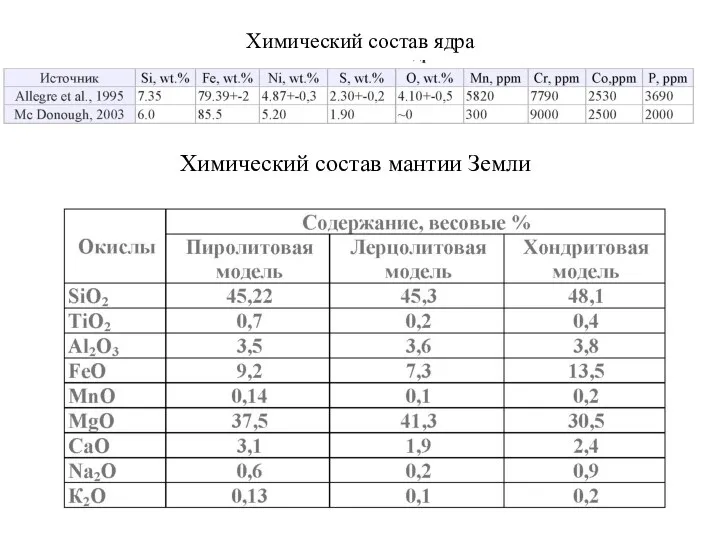
Химический состав ядра
Химический состав мантии Земли
Химический состав ядра
Химический состав мантии Земли

Лерцолит
Дунит
Лерцолит
Дунит

Полиморфизм
Впервые идея о возможности оливина под действием высоких давлений принимать структуру
Полиморфизм
Впервые идея о возможности оливина под действием высоких давлений принимать структуру
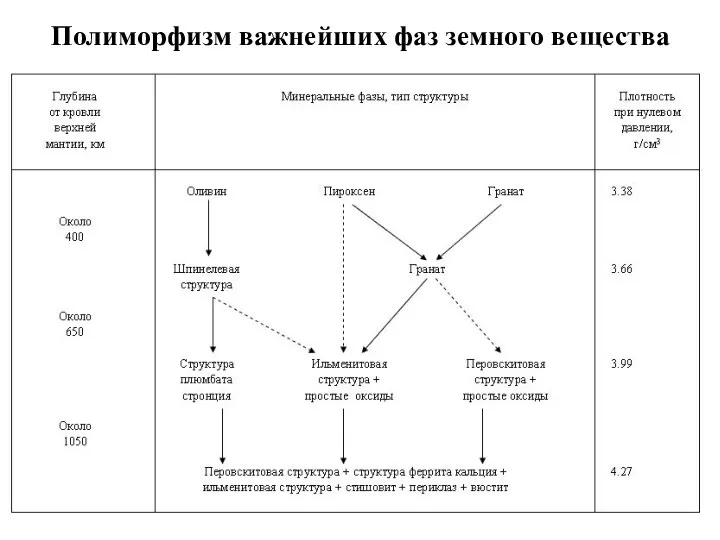
Полиморфизм важнейших фаз земного вещества
Полиморфизм важнейших фаз земного вещества

Шпинель
Гранат
Ильменит
Перовскит
Шпинель
Гранат
Ильменит
Перовскит

6. Распространенность химических элементов в верхней мантии Земли, оцениваемая на основании
6. Распространенность химических элементов в верхней мантии Земли, оцениваемая на основании
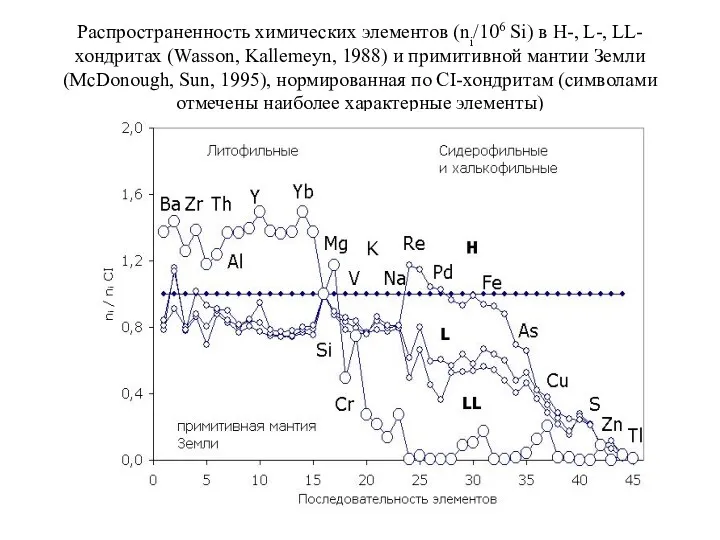
Распространенность химических элементов (ni/106 Si) в H-, L-, LL-хондритах (Wasson, Kallemeyn, 1988)
Распространенность химических элементов (ni/106 Si) в H-, L-, LL-хондритах (Wasson, Kallemeyn, 1988)

Состав вещества внутренних оболочек Земли
7. Изотопные данные заставляют предполагать, что
Состав вещества внутренних оболочек Земли
7. Изотопные данные заставляют предполагать, что
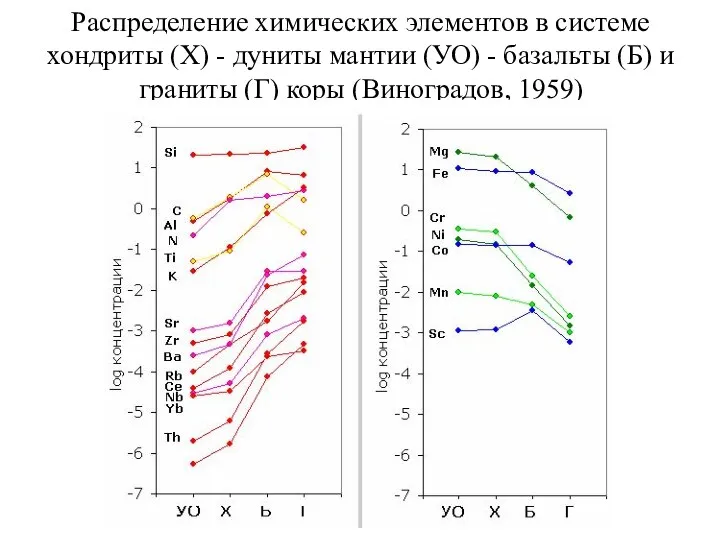
Распределение химических элементов в системе хондриты (Х) - дуниты мантии (УО)
Распределение химических элементов в системе хондриты (Х) - дуниты мантии (УО)

Из рассмотренного материала можно прийти к следующим заключениям:
В состав Земли
Из рассмотренного материала можно прийти к следующим заключениям:
В состав Земли

Основной химический состав геосфер
Основной химический состав геосфер

Порядки содержаний ХЭ в земной коре (г/т)
Порядки содержаний ХЭ в земной коре (г/т)
Лекция 5. Строение и химический состав земной коры
Лекция 5. Строение и химический состав земной коры

Скорость распространения сейсмических волн в континентальной земной коре и наиболее распространенных
Скорость распространения сейсмических волн в континентальной земной коре и наиболее распространенных
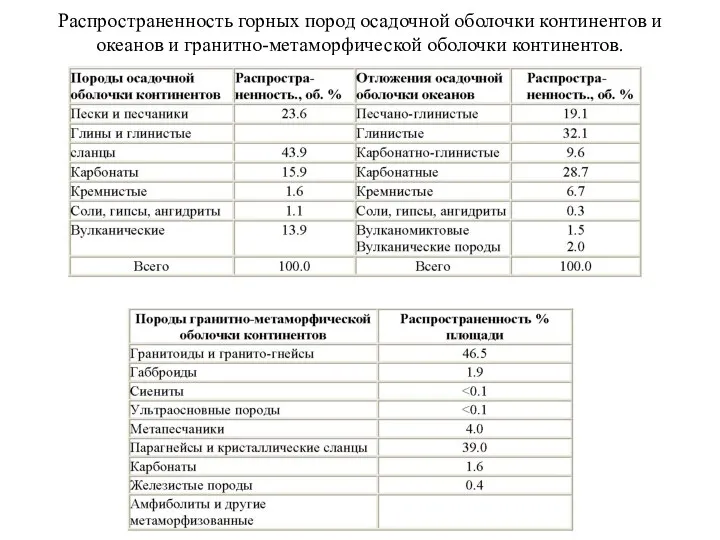
Распространенность горных пород осадочной оболочки континентов и океанов и гранитно-метаморфической оболочки
Распространенность горных пород осадочной оболочки континентов и океанов и гранитно-метаморфической оболочки

Земная кора
1. Средний химический состав земной коры отвечает средневзвешенному
Земная кора
1. Средний химический состав земной коры отвечает средневзвешенному

Распределение содержаний SiO2 в магматических породах (Richardson, Sneesby, 1923)
Распределение содержаний SiO2 в магматических породах (Richardson, Sneesby, 1923)

4. Статистика составов магматических пород позволила оценить относительную распространенность различных их
4. Статистика составов магматических пород позволила оценить относительную распространенность различных их

Земная кора
6. Альтернативные модели химического состава земной коры основаны на данных
Земная кора
6. Альтернативные модели химического состава земной коры основаны на данных

Земная кора
7. Состав гранулит-базитового слоя континентальной коры должен отличаться от состава
Земная кора
7. Состав гранулит-базитового слоя континентальной коры должен отличаться от состава

Земная кора
8. Оценка химического состава континентальной коры в целом остается неопределенной
Земная кора
8. Оценка химического состава континентальной коры в целом остается неопределенной
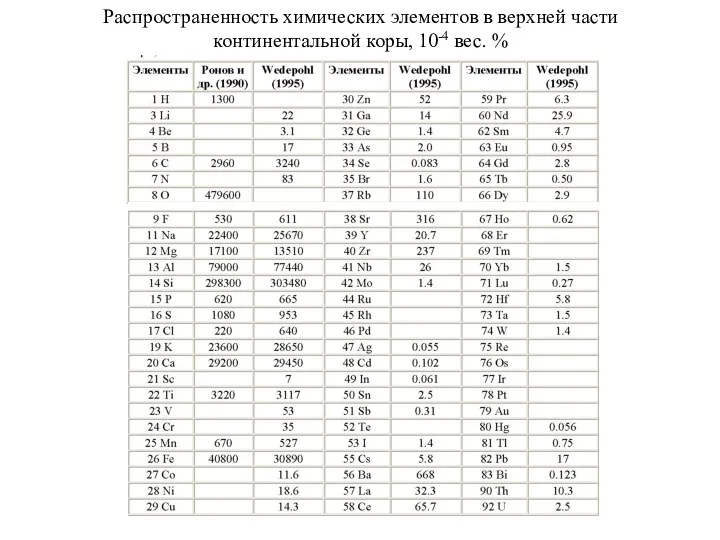
Распространенность химических элементов в верхней части континентальной коры, 10-4 вес. %
Распространенность химических элементов в верхней части континентальной коры, 10-4 вес. %

Земная кора
9. Средний химический состав гранитно-метаморфического слоя (верхней части континентальной коры)
Земная кора
9. Средний химический состав гранитно-метаморфического слоя (верхней части континентальной коры)

Земная кора
Континентальная кора существенно обогащена SiO2 и литофильными элементами с большим ионным
Земная кора
Континентальная кора существенно обогащена SiO2 и литофильными элементами с большим ионным

Земная кора
Выведенные на поверхность осадочные и магматич. породы подвергаются выветриванию – воздействию
Земная кора
Выведенные на поверхность осадочные и магматич. породы подвергаются выветриванию – воздействию

Лекция 6. Многообразие форм и видов нахождения химических элементов в природе
Лекция 6. Многообразие форм и видов нахождения химических элементов в природе

Минеральная форма.
В земной коре установлено более 4000 минеральных форм, что значительно
Минеральная форма.
В земной коре установлено более 4000 минеральных форм, что значительно
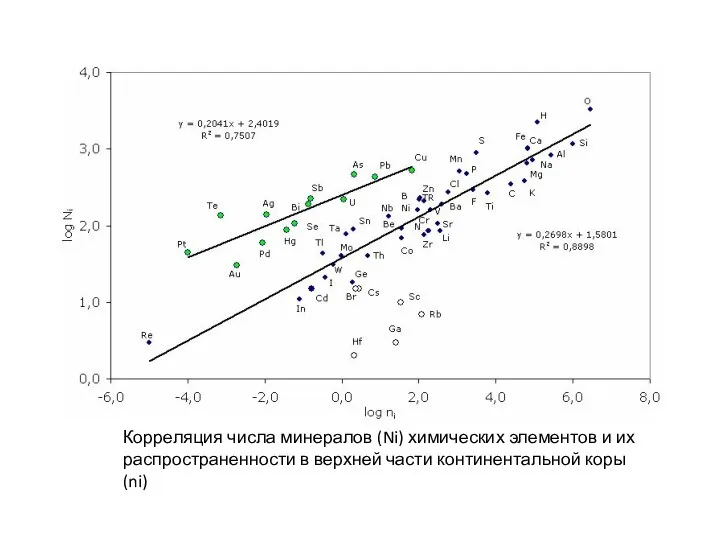
Корреляция числа минералов (Ni) химических элементов и их
распространенности в верхней части
Корреляция числа минералов (Ni) химических элементов и их
распространенности в верхней части

Способность химических элементов к минералообразованию
Способность химических элементов к минералообразованию
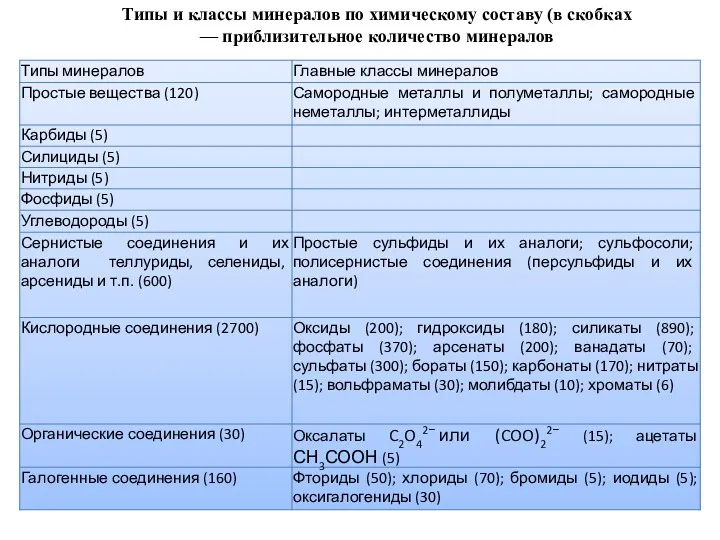
Типы и классы минералов по химическому составу (в скобках — приблизительное
Типы и классы минералов по химическому составу (в скобках — приблизительное

Формы нахождения химических элементов в рудах
Для основных рудных элементов характерна минеральная
Формы нахождения химических элементов в рудах
Для основных рудных элементов характерна минеральная

Участок Нембондачан (Au-Ag-полиметаллические руды)
Участок Нембондачан (Ag-полиметаллические руды)
Участок Нембондачан (полиметаллическая ассоциация)
Медные руды
Свинцовые
Участок Нембондачан (Au-Ag-полиметаллические руды)
Участок Нембондачан (Ag-полиметаллические руды)
Участок Нембондачан (полиметаллическая ассоциация)
Медные руды
Свинцовые

Т.н. 58/19 – образец жильного кварца с арсенопиритом и самородным золотом
Т.н. 58/19 – образец жильного кварца с арсенопиритом и самородным золотом

Агрегат лёллингита, арсенопирита, борнита, халькопирита и теннантита-тетраэдрита, обр. 72-31/5.
Участок Рыжий
Агрегат лёллингита, арсенопирита, борнита, халькопирита и теннантита-тетраэдрита, обр. 72-31/5.
Участок Рыжий

Самородное золото участка Туманный
золота
Теллуриды золота и серебра участка Вукней
Самородное золото участка Туманный
золота
Теллуриды золота и серебра участка Вукней

Образцы руд с сульфидно-сульфоантимонидной
минерализацией с участка Вернитакайвеем
Оруденелые метасоматиты
Кварцевые прожилки в
Образцы руд с сульфидно-сульфоантимонидной
минерализацией с участка Вернитакайвеем
Оруденелые метасоматиты
Кварцевые прожилки в
Серебро-полиметаллическая ассоциация участка Вернитакайвеем
Серебро-полиметаллическая ассоциация участка Вернитакайвеем

Золото-серебряная ассоциация
участка Вернитакайвеем
Россыпное золото
794%o
Plg Плагионит – Pb5Sb8S17
Zk Цинкенит – Pb6Sb14S27
Золото-серебряная ассоциация
участка Вернитакайвеем
Россыпное золото
794%o
Plg Плагионит – Pb5Sb8S17
Zk Цинкенит – Pb6Sb14S27

а) Вростки золота в пирите, б) электрум (пробность 684) в интерстициях
а) Вростки золота в пирите, б) электрум (пробность 684) в интерстициях
Ри
Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма
Ри
Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма
Ри
Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма
Ри
Серебросодержащие фазы. А) Вросток фазы Ag-As-Se-S в халькопирите. Б) кайма

Минералы карбонат-кварцевых жил. А, Б, Г) Гнезда карбонатов марганца (замещены оксидами
Минералы карбонат-кварцевых жил. А, Б, Г) Гнезда карбонатов марганца (замещены оксидами
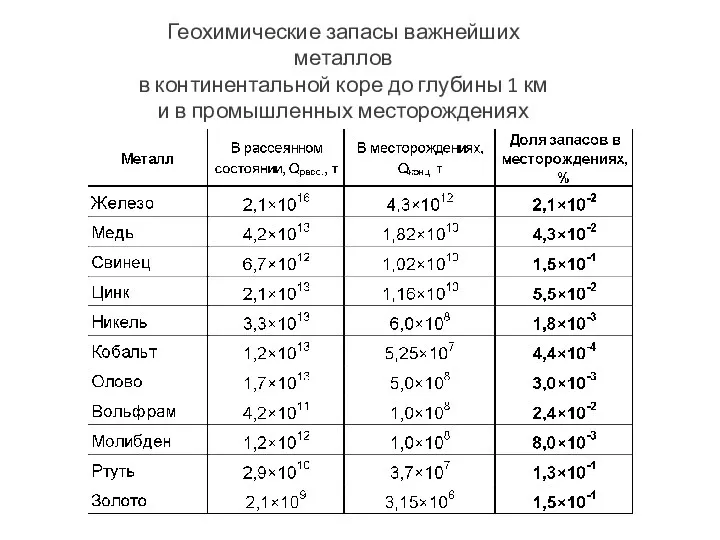
Геохимические запасы важнейших металлов
в континентальной коре до глубины 1 км
Геохимические запасы важнейших металлов
в континентальной коре до глубины 1 км
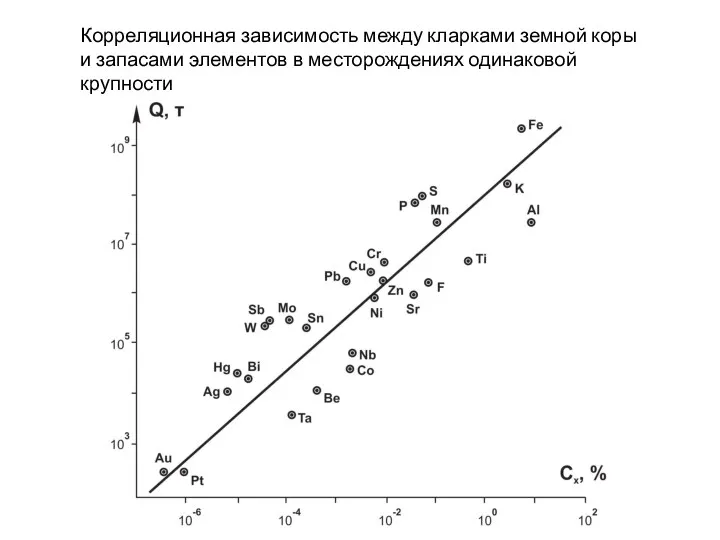
Корреляционная зависимость между кларками земной коры
и запасами элементов в месторождениях
Корреляционная зависимость между кларками земной коры
и запасами элементов в месторождениях
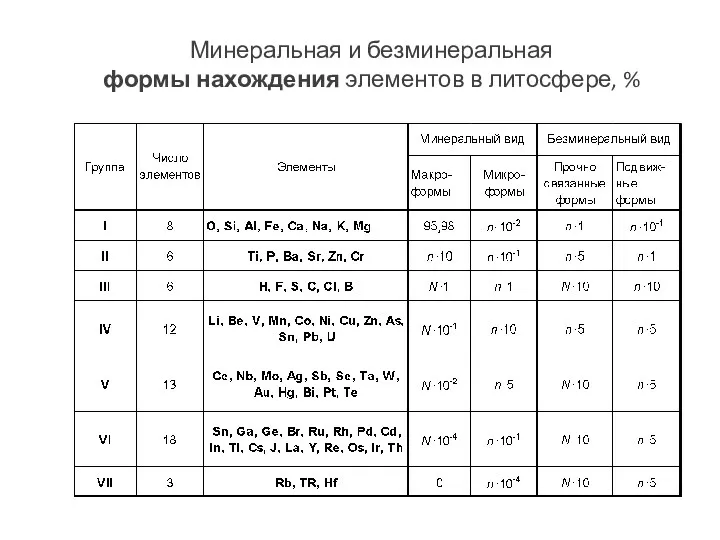
Минеральная и безминеральная
формы нахождения элементов в литосфере, %
Минеральная и безминеральная
формы нахождения элементов в литосфере, %

Безминеральная форма.
Наиболее характерными безминеральными формами нахождения элементов в природе являются следующие:
Безминеральная форма.
Наиболее характерными безминеральными формами нахождения элементов в природе являются следующие:

Лекция 7. Всеобщая миграция химических элементов во времени и пространстве
Космическим
Лекция 7. Всеобщая миграция химических элементов во времени и пространстве
Космическим

Миграция химических элементов
Находит отражение в гигантских тектоно-магматических процессах, преобразующих земную
Миграция химических элементов
Находит отражение в гигантских тектоно-магматических процессах, преобразующих земную
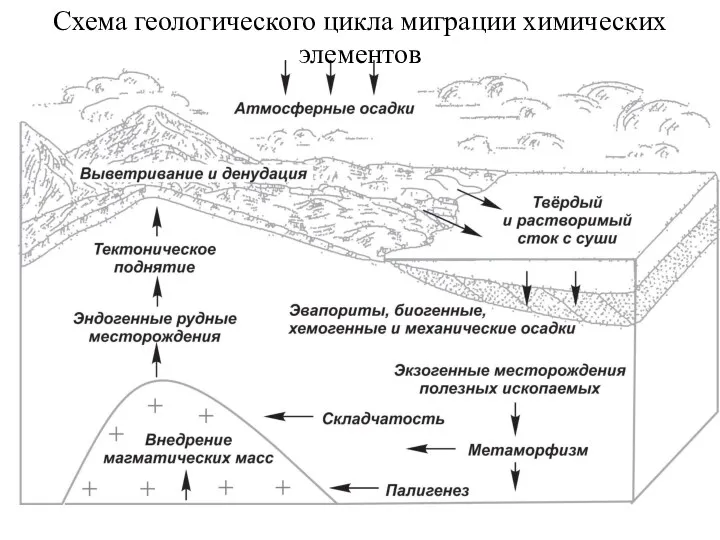
Схема геологического цикла миграции химических элементов
Схема геологического цикла миграции химических элементов

Существует два механизма переноса химических элементов в геологических процессах -
Существует два механизма переноса химических элементов в геологических процессах -

Конвективный массоперенос
Конвекция – физико-химическая миграция атомов, ионов, молекул вместе с растворителем.
Конвективный массоперенос
Конвекция – физико-химическая миграция атомов, ионов, молекул вместе с растворителем.

Диффузионный массоперенос
Диффузия – физико-химическая миграция вещества для установления равновесных концентраций вследствие
Диффузионный массоперенос
Диффузия – физико-химическая миграция вещества для установления равновесных концентраций вследствие

Гипогенная зона
характеризуется высокими и сверхвысокими температурами, давлением и концентрацией химических элементов,
Гипогенная зона
характеризуется высокими и сверхвысокими температурами, давлением и концентрацией химических элементов,

Гипергенная зона
Зона гипергенеза является главным местом действия солнечной радиации.
Под ее влиянием
Гипергенная зона
Зона гипергенеза является главным местом действия солнечной радиации.
Под ее влиянием

В магматических процессах основной вклад в перераспре-деление химических элементов вносит разделение
В магматических процессах основной вклад в перераспре-деление химических элементов вносит разделение
 Зеленая химия и проблемы устойчивого развития
Зеленая химия и проблемы устойчивого развития Аминокислоты. Пептиды. Хроматографические методы исследования
Аминокислоты. Пептиды. Хроматографические методы исследования The halogens
The halogens Материаловедение. Свойства материалов. (Тема 2)
Материаловедение. Свойства материалов. (Тема 2) Ароматичні вуглеводні (Арени). Бензен
Ароматичні вуглеводні (Арени). Бензен Крахмал. Строение вещества
Крахмал. Строение вещества Alkynes
Alkynes Химиялық реакциялардың типтері
Химиялық реакциялардың типтері Природные источники углеводородов
Природные источники углеводородов Топливо. Виды топлива
Топливо. Виды топлива p-элементы 17 группы периодической системы: галогены
p-элементы 17 группы периодической системы: галогены Создание пилотной производства 500 тонн в год рафинированного металлургического кремния Si 99,99% для нужд солнечной энергетики
Создание пилотной производства 500 тонн в год рафинированного металлургического кремния Si 99,99% для нужд солнечной энергетики Кристалічна ґрадка. Встановити взаємозв’язок між будовою речовин та їх фізичними властивостями
Кристалічна ґрадка. Встановити взаємозв’язок між будовою речовин та їх фізичними властивостями Ионные уравнения реакции
Ионные уравнения реакции Гидродинамические модели реакторов. Лекция № 2
Гидродинамические модели реакторов. Лекция № 2 Класифікація хімічних реакцій за кількістю і складом реагентів та продуктів реакції. 9 клас
Класифікація хімічних реакцій за кількістю і складом реагентів та продуктів реакції. 9 клас Органічні сполуки. 3D моделі органічних сполук
Органічні сполуки. 3D моделі органічних сполук Гидролиз. Классификация солей
Гидролиз. Классификация солей Аминокислоты. Номенклатура
Аминокислоты. Номенклатура Коррозия и методы борьбы с ней
Коррозия и методы борьбы с ней Managing chemicals. Green chemistry for every laboratory
Managing chemicals. Green chemistry for every laboratory Механохимиялық активтеу әдісі. Реакция түрлері
Механохимиялық активтеу әдісі. Реакция түрлері Heavy metals
Heavy metals Lipid metabolism
Lipid metabolism Основные разделы химии
Основные разделы химии Кислоты
Кислоты Химический элемент. Электронное строение атома
Химический элемент. Электронное строение атома Tungsten. (Вольфрам)
Tungsten. (Вольфрам)