- Общая и медицинская химия

Содержание
- 2. Рекомендуемая литература В нашей библиотеке: Хомченко И.Г. Общая химия, 1987 Глинка Н.Л. Общая химия, 1987 Глинка
- 3. Рекомендуемая литература Есть в мире: Общая и неорганическая химия для медиков и фармацевтов. Учебник и практикум
- 4. Периодический закон и периодическая система Основные понятия и закономерности
- 5. Периодическая система Графическое изображение Периодического закона на плоскости - периодическая система Наиболее распространенные формы периодической системы
- 6. Сверхдлинный вариант ПС
- 7. Длинный вариант ПС
- 8. Короткий вариант ПС
- 9. Развитие представлений о строении атома МОДЕЛИ АТОМОВ Кусочки материи –Демокрит полагал, что свойства того или иного
- 10. QUANTUM NUMBERS Quantum numbers: There are a set of four quantum numbers which specify the energy,
- 11. Principal quantum number (n) It determines the size and to a large extent the energy of
- 12. Azimuthal quantum number (l) It identified the sushell and the three dimensional shape of the orbital.
- 13. Magnetic quantum number or Magnetic orbital quantum number (ml) For any sub-shell (defined by ‘l’ value)
- 14. Shapes of Subshells
- 15. Filling of Electrons The filling of electrons into the orbitals of different atoms takes place according
- 16. Aufbau Principle: In the ground state of the atoms, the orbitals are filled in order of
- 17. Electronic configuration of atoms The electronic configuration of different atoms can be represented in two ways.
- 18. Electron spin quantum number (ms) It refers to orientation of the spin of the electron. It
- 19. Принципы и правила заполнения орбиталей Принцип Паули. В атоме не может быть двух электронов, у которых
- 20. Электронная конфигурация атома - запись, отражающая распределение электронов в атоме химического элемента по энергетическим уровням и
- 21. Правила написания электронных формул атомов Число энергетических уровней или значение главного квантового числа n равно номеру
- 22. Условная классификация простых веществ Металлы на внешнем уровне 1-3 электрона (искл.: Н, В, Не). характерна положительная
- 23. Основные характеристики химических элементов радиус атомов и ионов, энергия ионизации, энергия сродства к электрону, относительная электроотрицательность
- 24. Радиусы атомов и ионов Атомы и ионы не имеют точного размера (волновой характер), поэтому определяют условные
- 25. Зависимость атомного радиуса от заряда ядра
- 26. Зависимость атомного радиуса от заряда ядра По периоду. r уменьшается, т.к. при одинаковом значении n эффективный
- 27. Зависимость атомного радиуса от заряда ядра По подгруппам. В главной подгруппе r увеличивается сверху вниз, т.к.
- 28. Энергия ионизации атомов Энергия ионизации (I) - энергия, требуемая для отрыва и удаления на бесконечное расстояние
- 29. Периодичность энергии ионизации По периоду. Энергия ионизации растет, т.к. уменьшается радиус и растет эффективный заряд ядра.
- 30. Периодичность энергии ионизации По подгруппам наблюдается сложная зависимость, определяемая электронным строением и радиусом атомов. Радиус растет,
- 31. Энергия сродства к электрону Сродство к электрону определяется более всего энергией самой низкой незаполненной (или частично
- 32. Периодичность энергии сродства к электрону По периоду как правило энергия сродства к электрону увеличивается (кроме случая
- 33. Относительная электроотрицательность атомов Чем выше ЭО атома, тем в меньшей степени у него выражены восстановительные способности,
- 34. Относительная электроотрицательность атомов по Полингу По периоду Электроотрицательность увеличивается. По группам Электроотрицательность уменьшается Значения ЭО по
- 35. Практическая шкала электроотрицательности атомов В основу практической шкалы электроотрицательностей атомов взята концепция Луо-Бенсона, использующая понятие ковалентного
- 36. Относительная электроотрицательность атомов Элементу нельзя приписать постоянную электроотрицательность. На электроотрицательность оказывают влияние: Валентное состояния атома, Степень
- 37. Степени окисления Максимальная степень окисления в большинстве случаев равна номеру группы в короткопериодном варианте таблицы Менделеева.
- 38. Основы химической термодинамики Закономерности протекания химических реакций
- 39. Основные понятия химической термодинамики Задачи химической термодинамики: определение термодинамической вероятности протекания процесса определение термодинамических параметров процесса,
- 40. Основные понятия химической термодинамики Параметры – величины, характеризующие термодинамическую систему: интенсивные (величина которых не зависит от
- 41. Начала термодинамики Нулевое начало термодинамики: две системы, находящиеся в термическом равновесии с третьей системой, состоят в
- 42. Первое начало термодинамики внутренняя энергия системы U – определяется суммарным запасом энергии составляющих систему молекул, атомов,
- 43. Ни работа, ни теплота не являются функциями состояния и зависят от пути процесса (функции пути процесса).
- 44. Изохорный процесс V = const m = const Изотермический процесс T = const m = const
- 45. Первый закон термодинамики для изопроцессов
- 46. Термохимия Энтальпия – функция, характеризующая состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных
- 47. ЭНТРОПИЯ Энтропия - свойство системы, изменение которого при обратимом процессе равно отношению теплоты к температуре протекания
- 48. энтропия всегда увеличивается при переходе из конденсированного состояния в газообразное; она возрастает при растворении твердого или
- 49. Второе начало термодинамики в изолированных системах самопроизвольно идут процессы, при которых происходит увеличение энтропии.
- 50. Энергия Гиббса Любая система стремится к минимуму энтальпии и максимуму энтропии. В термодинамике имеется функция состояния,
- 51. Кинетика химических реакций Закономерности протекания химических реакций
- 52. Химическая кинетика изучает закономерности протекания химических реакций во времени, с ее помощью можно оптимизировать процессы и
- 53. Большинство химических реакций состоит из нескольких стадий, называемых элементарными реакциями. Под элементарной реакцией обычно понимают единичный
- 54. Молекулярность реакции Мономолекулярные реакции – элементарные реакции распада и изомеризации, в которых участвует только одна молекула
- 55. Скорость химической реакции Скорость химической реакции в газовой фазе или растворе определяется как изменениием числа молекул
- 56. Графический способ определения скорости химической реакции изменение концентрации одного из исходных веществ (1) и одного из
- 57. Факторы, влияющие на скорость химической реакции природа реагирующих веществ; концентрация реагирующих веществ; температура; наличие катализатора; величина
- 58. Формальная кинетика Для элементарных реакций константа скорости зависит только от температуры, а порядок по веществу совпадает
- 59. Кинетика реакций целого порядка Реакции 0-го порядка: скорость реакции не зависит от концентрации данного компонента при
- 60. Реакции 1-го порядка: реакции типа A → С. скорость прямо пропорциональна концентрации период полураспада τ1/2.
- 61. Реакции 2-го порядка: скорость прямо пропорциональна произведению концентраций При решении этого уравнения различают два случая: Одинаковые
- 62. Реакции 3-го порядка А+В+С → продукты или 2А+В → продукты, 3А → продукты и т.д.
- 63. Реакции n-го порядка А+В+С......→ продукты реакции
- 64. Метод определения порядка реакции интегральные дифференциальные используют интегральные кинетические уравнения для обработки экспериментальных данных о зависимости
- 65. Метод подстановки (метод проб и ошибок) Экспериментальные данные последовательно подставляют в интегральные кинетические уравнения для реакций
- 66. Метод Раковского Изучают зависимость периода полупревращения от С0. Для реакций первого порядка t1/2 не зависит от
- 67. Метод Вант-Гоффа Аналитический Графический Cтроят зависимость lnυ от ln[A]. Из тангенса угла наклона полученной прямой определяют
- 68. Метод изолирования Оствальда зависимость скорости реакции от начальной концентрации одного из реагентов (например, A) изучают при
- 69. Зависимость скорости реакции от температуры правило ВантГоффа: при увеличении температуры на каждые 10 градусов скорость возрастает
- 70. Химическая связь. Гибридизация.
- 71. Химическая связь Внешние оболочки всех элементов, кроме благородных газов, являются НЕЗАВЕРШЕННЫМИ и в процессе химического взаимодействия
- 72. Ковалентная связь Образование может происходить двумя путями: Коллигация 2. Координация (донорно-акцепторная связь) Химическая связь, образованная путем
- 73. Ковалентная неполярная связь атомы образуют химические связи в результате обобществ-ления такого количества электронов, чтобы приобрести электронную
- 74. Ковалентная полярная связь Мерой полярности связи является ее дипольный момент μ : μ = е l,
- 75. Ионная связь Ионная химическая связь представляет собой электростатическое взаимодействие положительных и отрицательных заряженных ионов в химическом
- 76. Полярность связи Для чисто ковалентной связи Δ X = 0. Если величина Δ X меньше, чем
- 77. Металлическая связь характерна только для металлов: атомы металлов имеют большое число валентных атомных орбиталей и недостаток
- 78. Водородная связь Водородная связь по природе относится к электростатическим и образуется в том случае, когда атом
- 79. Примеры водородной связи Спираль ДНК Белки Структура воды
- 80. Характеристики химической связи Полярность связи - степень смещения электронной плотности, приводящего к возникновению разнополярных полюсов. Молекула
- 81. Метод валентных связей (МВС) Ковалентную химическую связь образуют два электрона с противоположными спинами, принадлежащие двум атомам.
- 82. Метод молекулярных орбиталей (ММО) состояние электронов в молекуле может быть описано как совокупность молекулярных электронных орбиталей,
- 83. Кратные связи делокализованная π-связь метод наложения валентных схем на примере азидоводорода HN3 в молекуле остаются два
- 85. Скачать презентацию

Рекомендуемая литература
В нашей библиотеке:
Хомченко И.Г. Общая химия, 1987
Глинка Н.Л. Общая химия,
Рекомендуемая литература
В нашей библиотеке:
Хомченко И.Г. Общая химия, 1987
Глинка Н.Л. Общая химия,

Рекомендуемая литература
Есть в мире:
Общая и неорганическая химия для медиков и фармацевтов.
Рекомендуемая литература
Есть в мире:
Общая и неорганическая химия для медиков и фармацевтов.

Периодический закон и периодическая система
Основные понятия и закономерности
Периодический закон и периодическая система
Основные понятия и закономерности

Периодическая система
Графическое изображение Периодического закона на плоскости - периодическая система
Наиболее
Периодическая система
Графическое изображение Периодического закона на плоскости - периодическая система
Наиболее

Сверхдлинный вариант ПС
Сверхдлинный вариант ПС

Длинный вариант ПС
Длинный вариант ПС
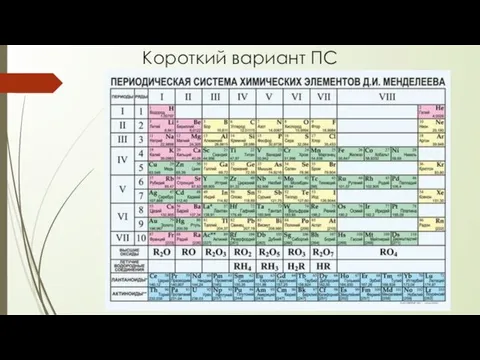
Короткий вариант ПС
Короткий вариант ПС

Развитие представлений о строении атома
МОДЕЛИ АТОМОВ
Кусочки материи –Демокрит полагал, что свойства того
Развитие представлений о строении атома
МОДЕЛИ АТОМОВ
Кусочки материи –Демокрит полагал, что свойства того

QUANTUM NUMBERS
Quantum numbers: There are a set of four quantum numbers
QUANTUM NUMBERS
Quantum numbers: There are a set of four quantum numbers

Principal quantum number (n)
It determines the size and to a large
Principal quantum number (n)
It determines the size and to a large

Azimuthal quantum number (l)
It identified the sushell and the three dimensional
Azimuthal quantum number (l)
It identified the sushell and the three dimensional

Magnetic quantum number or Magnetic orbital quantum number (ml)
For any sub-shell
Magnetic quantum number or Magnetic orbital quantum number (ml)
For any sub-shell
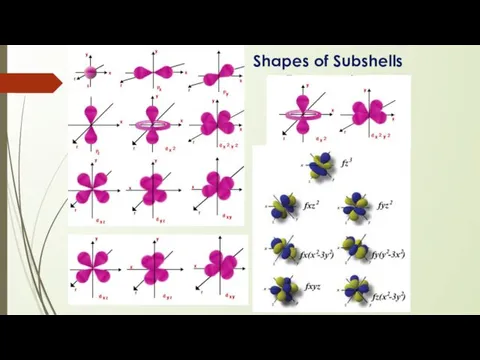
Shapes of Subshells
Shapes of Subshells

Filling of Electrons
The filling of electrons into the orbitals of different
Filling of Electrons
The filling of electrons into the orbitals of different

Aufbau Principle: In the ground state of the atoms, the
Aufbau Principle: In the ground state of the atoms, the

Electronic configuration of atoms
The electronic configuration of different atoms can be
Electronic configuration of atoms
The electronic configuration of different atoms can be

Electron spin quantum number (ms)
It refers to orientation of the spin
Electron spin quantum number (ms)
It refers to orientation of the spin

Принципы и правила заполнения орбиталей
Принцип Паули. В атоме не может быть
Принципы и правила заполнения орбиталей
Принцип Паули. В атоме не может быть

Электронная конфигурация атома - запись, отражающая распределение электронов в атоме химического
Электронная конфигурация атома - запись, отражающая распределение электронов в атоме химического

Правила написания электронных формул атомов
Число энергетических уровней или значение главного квантового
Правила написания электронных формул атомов
Число энергетических уровней или значение главного квантового

Условная классификация простых веществ
Металлы
на внешнем уровне 1-3 электрона (искл.: Н, В,
Условная классификация простых веществ
Металлы
на внешнем уровне 1-3 электрона (искл.: Н, В,

Основные характеристики химических элементов
радиус атомов и ионов,
энергия ионизации,
энергия сродства
Основные характеристики химических элементов
радиус атомов и ионов,
энергия ионизации,
энергия сродства

Радиусы атомов и ионов
Атомы и ионы не имеют точного размера (волновой
Радиусы атомов и ионов
Атомы и ионы не имеют точного размера (волновой
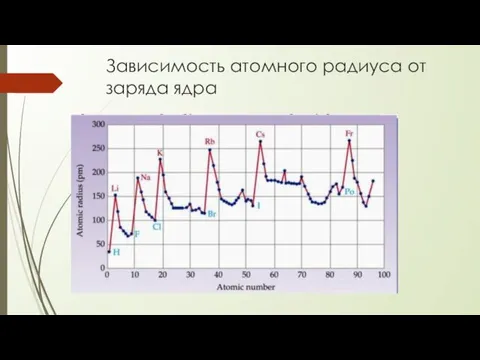
Зависимость атомного радиуса от заряда ядра
Зависимость атомного радиуса от заряда ядра

Зависимость атомного радиуса от заряда ядра
По периоду.
r уменьшается, т.к.
Зависимость атомного радиуса от заряда ядра
По периоду.
r уменьшается, т.к.

Зависимость атомного радиуса от заряда ядра
По подгруппам.
В главной подгруппе
Зависимость атомного радиуса от заряда ядра
По подгруппам.
В главной подгруппе

Энергия ионизации атомов
Энергия ионизации (I) - энергия, требуемая для отрыва
Энергия ионизации атомов
Энергия ионизации (I) - энергия, требуемая для отрыва

Периодичность энергии ионизации
По периоду.
Энергия ионизации растет, т.к. уменьшается
Периодичность энергии ионизации
По периоду.
Энергия ионизации растет, т.к. уменьшается

Периодичность энергии ионизации
По подгруппам
наблюдается сложная зависимость, определяемая электронным строением
Периодичность энергии ионизации
По подгруппам
наблюдается сложная зависимость, определяемая электронным строением

Энергия сродства к электрону
Сродство к электрону определяется более всего энергией самой
Энергия сродства к электрону
Сродство к электрону определяется более всего энергией самой

Периодичность энергии сродства к электрону
По периоду
как правило энергия сродства к электрону
Периодичность энергии сродства к электрону
По периоду
как правило энергия сродства к электрону

Относительная электроотрицательность атомов
Чем выше ЭО атома, тем в меньшей степени у
Относительная электроотрицательность атомов
Чем выше ЭО атома, тем в меньшей степени у

Относительная электроотрицательность атомов по Полингу
По периоду
Электроотрицательность увеличивается.
По группам
Электроотрицательность уменьшается
Значения ЭО
Относительная электроотрицательность атомов по Полингу
По периоду
Электроотрицательность увеличивается.
По группам
Электроотрицательность уменьшается
Значения ЭО

Практическая шкала электроотрицательности атомов
В основу практической шкалы электроотрицательностей атомов взята концепция
Практическая шкала электроотрицательности атомов
В основу практической шкалы электроотрицательностей атомов взята концепция

Относительная электроотрицательность атомов
Элементу нельзя приписать постоянную электроотрицательность.
На электроотрицательность оказывают влияние:
Валентное
Относительная электроотрицательность атомов
Элементу нельзя приписать постоянную электроотрицательность.
На электроотрицательность оказывают влияние:
Валентное

Степени окисления
Максимальная степень окисления в большинстве случаев равна номеру группы в
Степени окисления
Максимальная степень окисления в большинстве случаев равна номеру группы в

Основы химической термодинамики
Закономерности протекания химических реакций
Основы химической термодинамики
Закономерности протекания химических реакций

Основные понятия химической термодинамики
Задачи химической термодинамики:
определение термодинамической вероятности протекания процесса
Основные понятия химической термодинамики
Задачи химической термодинамики:
определение термодинамической вероятности протекания процесса

Основные понятия химической термодинамики
Параметры – величины, характеризующие термодинамическую систему:
интенсивные (величина
Основные понятия химической термодинамики
Параметры – величины, характеризующие термодинамическую систему:
интенсивные (величина

Начала термодинамики
Нулевое начало термодинамики:
две системы, находящиеся в термическом равновесии с третьей
Начала термодинамики
Нулевое начало термодинамики:
две системы, находящиеся в термическом равновесии с третьей

Первое начало термодинамики
внутренняя энергия системы U – определяется суммарным запасом энергии
Первое начало термодинамики
внутренняя энергия системы U – определяется суммарным запасом энергии

Ни работа, ни теплота не являются функциями состояния и зависят от
Ни работа, ни теплота не являются функциями состояния и зависят от

Изохорный процесс
V = const
m = const
Изотермический процесс
T = const
m = const
Изобарный
Изохорный процесс
V = const
m = const
Изотермический процесс
T = const
m = const
Изобарный

Первый закон термодинамики для изопроцессов
Первый закон термодинамики для изопроцессов

Термохимия
Энтальпия – функция, характеризующая состояние системы в термодинамическом равновесии при
Термохимия
Энтальпия – функция, характеризующая состояние системы в термодинамическом равновесии при

ЭНТРОПИЯ
Энтропия - свойство системы, изменение которого при обратимом процессе равно отношению
ЭНТРОПИЯ
Энтропия - свойство системы, изменение которого при обратимом процессе равно отношению

энтропия всегда увеличивается при переходе из конденсированного состояния в газообразное;
она
энтропия всегда увеличивается при переходе из конденсированного состояния в газообразное;
она

Второе начало термодинамики
в изолированных системах самопроизвольно идут процессы, при которых
Второе начало термодинамики
в изолированных системах самопроизвольно идут процессы, при которых

Энергия Гиббса
Любая система стремится к минимуму энтальпии и максимуму энтропии.
В
Энергия Гиббса
Любая система стремится к минимуму энтальпии и максимуму энтропии.
В

Кинетика
химических реакций
Закономерности протекания химических реакций
Кинетика
химических реакций
Закономерности протекания химических реакций
Химическая кинетика изучает закономерности протекания химических реакций во времени, с ее
Химическая кинетика изучает закономерности протекания химических реакций во времени, с ее

Большинство химических реакций состоит из нескольких стадий, называемых элементарными реакциями.
Под элементарной
Большинство химических реакций состоит из нескольких стадий, называемых элементарными реакциями.
Под элементарной

Молекулярность реакции
Мономолекулярные реакции – элементарные реакции распада и изомеризации, в которых
Молекулярность реакции
Мономолекулярные реакции – элементарные реакции распада и изомеризации, в которых

Скорость химической реакции
Скорость химической реакции в газовой фазе или растворе определяется
Скорость химической реакции
Скорость химической реакции в газовой фазе или растворе определяется
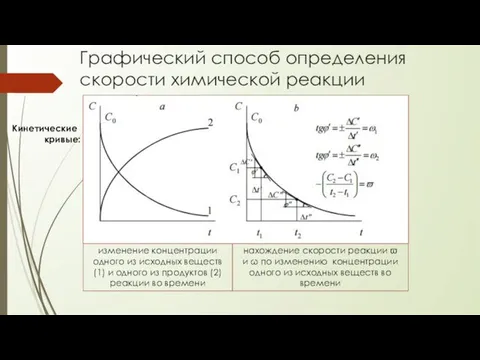
Графический способ определения скорости химической реакции
изменение концентрации одного из исходных веществ
Графический способ определения скорости химической реакции
изменение концентрации одного из исходных веществ

Факторы, влияющие на скорость химической реакции
природа реагирующих веществ;
концентрация реагирующих веществ;
Факторы, влияющие на скорость химической реакции
природа реагирующих веществ;
концентрация реагирующих веществ;

Формальная кинетика
Для элементарных реакций константа скорости зависит только от температуры, а
Формальная кинетика
Для элементарных реакций константа скорости зависит только от температуры, а

Кинетика реакций целого порядка
Реакции 0-го порядка:
скорость реакции не зависит от
Кинетика реакций целого порядка
Реакции 0-го порядка:
скорость реакции не зависит от
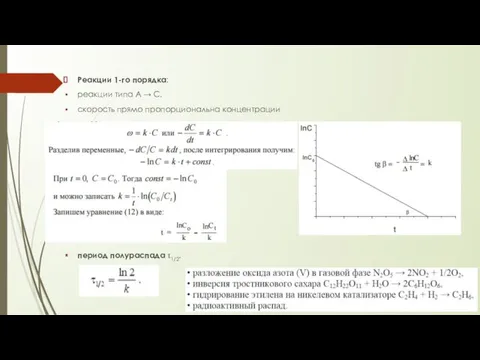
Реакции 1-го порядка:
реакции типа A → С.
скорость прямо пропорциональна
Реакции 1-го порядка:
реакции типа A → С.
скорость прямо пропорциональна
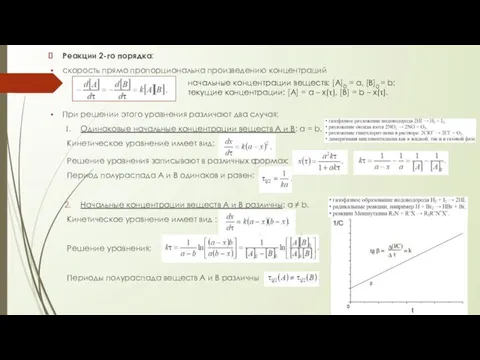
Реакции 2-го порядка:
скорость прямо пропорциональна произведению концентраций
При решении этого уравнения
Реакции 2-го порядка:
скорость прямо пропорциональна произведению концентраций
При решении этого уравнения

Реакции 3-го порядка
А+В+С → продукты или 2А+В → продукты, 3А
Реакции 3-го порядка
А+В+С → продукты или 2А+В → продукты, 3А

Реакции n-го порядка
А+В+С......→ продукты реакции
Реакции n-го порядка
А+В+С......→ продукты реакции

Метод определения порядка реакции
интегральные
дифференциальные
используют интегральные кинетические уравнения для обработки экспериментальных данных
Метод определения порядка реакции
интегральные
дифференциальные
используют интегральные кинетические уравнения для обработки экспериментальных данных

Метод подстановки (метод проб и ошибок)
Экспериментальные данные последовательно подставляют в интегральные
Метод подстановки (метод проб и ошибок)
Экспериментальные данные последовательно подставляют в интегральные
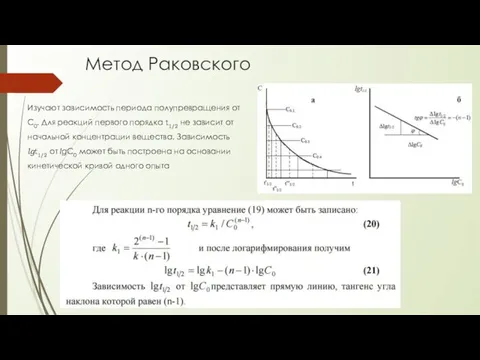
Метод Раковского
Изучают зависимость периода полупревращения от С0. Для реакций первого порядка
Метод Раковского
Изучают зависимость периода полупревращения от С0. Для реакций первого порядка
![Метод Вант-Гоффа Аналитический Графический Cтроят зависимость lnυ от ln[A]. Из](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/108066/slide-66.jpg)
Метод Вант-Гоффа
Аналитический
Графический
Cтроят зависимость lnυ от ln[A].
Из тангенса угла наклона полученной
Метод Вант-Гоффа
Аналитический
Графический
Cтроят зависимость lnυ от ln[A].
Из тангенса угла наклона полученной

Метод изолирования Оствальда
зависимость скорости реакции от начальной концентрации одного из реагентов
Метод изолирования Оствальда
зависимость скорости реакции от начальной концентрации одного из реагентов

Зависимость скорости реакции от температуры
правило ВантГоффа: при увеличении температуры на
Зависимость скорости реакции от температуры
правило ВантГоффа: при увеличении температуры на

Химическая связь. Гибридизация.
Химическая связь. Гибридизация.
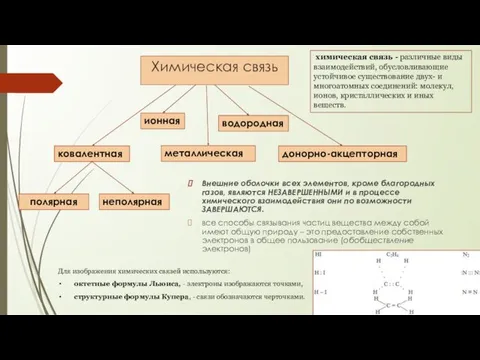
Химическая связь
Внешние оболочки всех элементов, кроме благородных газов, являются НЕЗАВЕРШЕННЫМИ и
Химическая связь
Внешние оболочки всех элементов, кроме благородных газов, являются НЕЗАВЕРШЕННЫМИ и

Ковалентная связь
Образование может происходить двумя путями:
Коллигация
2. Координация
(донорно-акцепторная
связь)
Химическая связь, образованная
Ковалентная связь
Образование может происходить двумя путями:
Коллигация
2. Координация
(донорно-акцепторная
связь)
Химическая связь, образованная

Ковалентная неполярная связь
атомы образуют химические связи в результате обобществ-ления такого количества
Ковалентная неполярная связь
атомы образуют химические связи в результате обобществ-ления такого количества

Ковалентная полярная связь
Мерой полярности связи является ее дипольный момент μ :
μ
Ковалентная полярная связь
Мерой полярности связи является ее дипольный момент μ :
μ

Ионная связь
Ионная химическая связь представляет собой электростатическое взаимодействие положительных и отрицательных заряженных
Ионная связь
Ионная химическая связь представляет собой электростатическое взаимодействие положительных и отрицательных заряженных

Полярность связи
Для чисто ковалентной связи Δ X = 0.
Если величина Δ X меньше,
Полярность связи
Для чисто ковалентной связи Δ X = 0.
Если величина Δ X меньше,
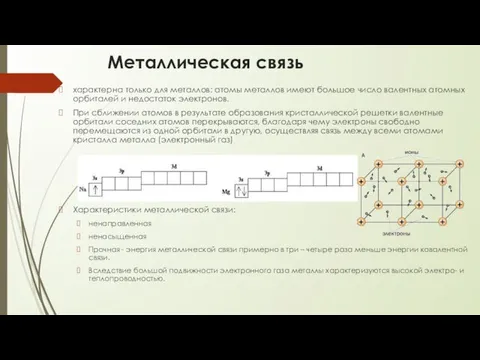
Металлическая связь
характерна только для металлов: атомы металлов имеют большое число валентных
Металлическая связь
характерна только для металлов: атомы металлов имеют большое число валентных

Водородная связь
Водородная связь по природе относится к электростатическим и образуется в
Водородная связь
Водородная связь по природе относится к электростатическим и образуется в
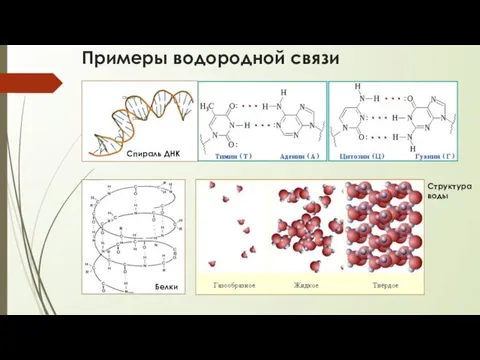
Примеры водородной связи
Спираль ДНК
Белки
Структура
воды
Примеры водородной связи
Спираль ДНК
Белки
Структура
воды

Характеристики химической связи
Полярность связи - степень смещения электронной плотности, приводящего к возникновению
Характеристики химической связи
Полярность связи - степень смещения электронной плотности, приводящего к возникновению

Метод валентных связей (МВС)
Ковалентную химическую связь образуют два электрона с противоположными спинами,
Метод валентных связей (МВС)
Ковалентную химическую связь образуют два электрона с противоположными спинами,

Метод молекулярных орбиталей (ММО)
состояние электронов в молекуле может быть описано как
Метод молекулярных орбиталей (ММО)
состояние электронов в молекуле может быть описано как

Кратные связи делокализованная π-связь
метод наложения валентных схем на примере азидоводорода HN3
в
Кратные связи делокализованная π-связь
метод наложения валентных схем на примере азидоводорода HN3
в
 Композиционные материалы для изоляции электрических машин
Композиционные материалы для изоляции электрических машин Кислоты НСL, H2 O, H2 CO3
Кислоты НСL, H2 O, H2 CO3 Физические явления – основа разделения смесей в химии (урок химии в 8 классе)
Физические явления – основа разделения смесей в химии (урок химии в 8 классе) Зиянды организмдерге қарсы органикалық және органикалық емес қосылыстарды қолдануға негізделген тәсіл
Зиянды организмдерге қарсы органикалық және органикалық емес қосылыстарды қолдануға негізделген тәсіл Цинк и его применение
Цинк и его применение Полимеры. Каучук
Полимеры. Каучук Полисахариды. Крахмал
Полисахариды. Крахмал Третья группа, главная подгруппа. 9 класс
Третья группа, главная подгруппа. 9 класс Сложные эфиры
Сложные эфиры Закон сохранения массы веществ. Уравнения химических реакций
Закон сохранения массы веществ. Уравнения химических реакций Значення хімічних процесів у природі
Значення хімічних процесів у природі Курс лекций: Методы диагностики и анализа микро- и наносистем
Курс лекций: Методы диагностики и анализа микро- и наносистем Твердое состояние вещества. Кристаллические и аморфные тела
Твердое состояние вещества. Кристаллические и аморфные тела Гидроксиды. Основания: способы получения
Гидроксиды. Основания: способы получения Жер қыртысының заттық және химиялық құрамы
Жер қыртысының заттық және химиялық құрамы Реакции деструкции макромолекул
Реакции деструкции макромолекул Неметаллы
Неметаллы Окислительно-восстановительные реакции
Окислительно-восстановительные реакции Классификация веществ по характеру связи
Классификация веществ по характеру связи Распределение электронов в атомах. 8 класс
Распределение электронов в атомах. 8 класс Chemistry of Coordination Compounds
Chemistry of Coordination Compounds Общая электронная теория восстановления и окисления металлов
Общая электронная теория восстановления и окисления металлов Субстраты и продукты биохимических реакций
Субстраты и продукты биохимических реакций Пурины. Строение пурина
Пурины. Строение пурина Высокомолекулярные соединения полимеры
Высокомолекулярные соединения полимеры Химическая связь
Химическая связь Понятия про синтетические лекарственные средства
Понятия про синтетические лекарственные средства Получение и применение алканов
Получение и применение алканов