- Промышленные катализаторы

Содержание
- 2. Катализаторы стратегическая продукция Малым количеством катализатора можно превратить громадные количества вещества (фактор использования вещества катализатора ~104-106)
- 3. Промышленные российские технологии каталитических процессов
- 4. Нефтехимия России 16
- 5. Катализаторы стратегическая продукция Объем мирового рынка катализаторов - 17,5 млрд.долл.США/год. В России 15 млрд.рублей/год Темпы ежегодного
- 6. Катализаторы стратегическая продукция Зависимость отечественной технологии от поставок зарубежных катализаторов: - Каталитический крекинг на 80 %
- 7. Катализаторы стратегическая продукция Анализ показывает масштабный спад в областях разработки, обновления ассортимента и модернизации производства катализаторов.
- 8. Катализаторы стратегическая продукция Причины Наукоемкость ОТСТАВАНИЕ РОССИЙСКИХ РАЗРАБОТОК ВО МНОГИХ ОБЛАСТЯХ КАТАЛИТИЧЕСКИХ ТЕХНОЛОГИЙ Плохая наследственность по
- 9. Катализаторы стратегическая продукция Пути решения Создание условий для приоритетного использования отечественных НТР в области катализа; Создание
- 10. РОССИЙСКИЕ ПРОИЗВОДИТЕЛИ КАТАЛИЗАТОРОВ
- 11. Прогнозируемая годовая потребность России в катализаторах гидроочистки Современные промышленные катализаторы гидроочистки Нанесённые зарубежные катализаторы Haldor Topsoe,
- 12. Современное состояние и направления развития катализаторов гидроочистки
- 13. Лекции 1, 2 Исторические корни катализа XV век: алхимики 1669 г.: Бекер 1759 г.: Шееле, Бухольц
- 14. Лекции 1, 2 Исторические корни катализа 1806 г.: Клеман, Дезорм 1811 г.: Кирхгоф 2SO2 + O2
- 15. Лекции 1, 2 Исторические корни катализа 1813 г.: Тенар 1817 г.: Дэви 1818 г.: Тенар 1821
- 16. Лекции 1, 2 Становление катализа 1834 г.: Митчерлих ввел понятие «контактные реакции» 1835 г.: Берцелиус ввел
- 17. Лекции 1, 2 Основные положения определения катализа À Возбуждение химических реакций (а не только ускорение) Á
- 18. Лекции 1, 2 Основные положения определения катализа
- 19. Лекции 1, 2 Роль катализа в становлении и развитии современной промышленности 1875 г.: Окисление SO2 в
- 20. Лекции 1, 2 Роль катализа в становлении и развитии современной промышленности 1930-1932 г.г.: Синтез искусственного каучука
- 21. Лекции 1, 2 Роль катализа в становлении и развитии современной промышленности 1950-1960 г.г.: Процессы гидроочистки —
- 22. Лекции 1, 2 Катализ в решении проблем энергетики 1. Каталитическое сжигание топлив
- 23. Лекции 1, 2 Катализ в решении проблем энергетики 2. Запасание солнечной энергии а) термокаталитически б) фотокаталитически
- 24. Лекции 1, 2 Катализ в решении проблем энергетики 3. Создание топливных элементов (к.п.д. – 70%) 4.
- 25. Лекции 1, 2 Катализ в решении проблем экологии топливо воздух сточные воды (фенол, анилин) CO2, N2,
- 26. Лекции 1, 2 Катализ в живой природе Почти все реакции в клетках живых организмов — каталитические.
- 27. Лекции 1, 2 Катализ в живой природе И.П.Павлов назвал ферменты “возбудителями жизни“ Число найденных ферментов >2000;
- 28. Лекции 1, 2 Классификация катализаторов À Катализатор может быть как индивидуальным веществом, так и смесью веществ
- 29. Лекции 1, 2 Классификация катализаторов Â Катализатор может быть массивным, нанесенным и закрепленным Понятие об активном
- 30. Лекции 1, 2 Классификация катализаторов Массивный катализатор целиком состоит из активного компонента Нанесенный катализатор: активный компонент
- 31. Лекции 1, 2 Классификация каталитических процессов À Гомогенный катализ Â Ферментативный катализ
- 32. Лекции 1, 2 Понятие о каталитическом цикле Каталитический цикл — система реакций с участием катализатора, при
- 33. Лекции 1, 2 Каталитическая активность число оборотов
- 34. Лекции 1, 2 Селективность каталитического процесса
- 35. Лекции 1, 2
- 36. Лекции 1, 2 Для рентабельной эксплуатации катализатор должен обладать 1.Высокой каталитической активностью и селективностью; 2.Достаточно развитой
- 37. Лекции 1, 2 Стабильность К числу главных характеристик катализатора относится его устойчивость к длительной работе, которая
- 38. Лекции 1, 2 дисперсность Термин дисперсность – производный от лат. слова dispersus (рассеянный, рассыпанный), в современной
- 39. Лекции 1, 2 Механическая прочность Прочность тонкодисперсного твердого тела в значительной степени зависит не от прочности
- 40. Лекции 1, 2 Определение и характеристики пористых материалов. Мы знаем, что существуют понятия истинной и кажущейся
- 41. Лекции 1, 2 Супрамолекулярная структура пористого материала. Пористые материалы характеризуются величинами удельной поверхности, пористости, кажущейся плотности,
- 42. Лекции 1, 2 классификации пористых тел по размерам пор Действительно, каково бы ни было строение пористых
- 43. Лекции 1, 2 Морфология пористых тел
- 44. Лекции 1, 2 СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ Известны цеолиты различных типов. Тип, к которому относится данный цеолит, зависит
- 45. Лекции 1, 2 СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ [Мn]+ [(Al2O3)m×(SiO2)k×(H2O)l]- [Мn]+= H+, K+ , Na+, Ca2+…. SiO2/Al2O3 тип цеолита
- 46. Объемы производства Мировое –1,5 млн. тонн в год (~5 млрд. $); Россия – 7 тыс. тонн
- 47. Лекции 1, 2
- 48. Схема комплексного подхода к решению основ промышленного производства катализаторов. Лекции 1, 2
- 49. Лекции 1, 2 Традиционные методы синтеза пористых материалов - катализаторов 1. Осаждение (соосаждение для многокомпонентных систем)
- 50. Лекции 1, 2 Основные параметры и факторы осаждения
- 51. Лекции 1, 2 . Технологии приготовления никелевых катализаторов гидрирования
- 53. Восстановительное разложение основного карбоната никеля (активация)
- 54. Технология приготовления катализаторов гидрирования методами осаждения через аммиачно-карбонатные комплексы
- 55. Технология приготовления. Алюмо-цинк-медные катализаторы гидрирования 1. Приготовление аммиачно-карбонатного комплекса меди Карбонатный медно-аммиачный раствор готовят растворением гидрокарбоната
- 56. Лекции 1, 2 Физико-химические основы получения пористых материалов методами нанесения Требования, предъявляемые к носителям Носитель должен
- 57. Лекции 1, 2 Способы нанесения Состояние и дисперсность активного компонента в нанесенных катализаторах зависит от большого
- 58. Лекции 1, 2 Режимы пропитки Капиллярная пропитка. Нанесение вещества осуществляется за счет всасывания раствора в поры
- 59. раствор КОН H2CrO4 Носитель
- 60. раствор КОН H2CrO4 Носитель Сушка Т = 120 ±10 оС Пропитка Т = 40 – 60
- 61. Схема приготовления катализатора
- 62. Способы формовки Таблетирование (штамбовка) формование порошков методом таблетирования Преимущество: строго определенный длины Недостатки: Дорогостоящее прецизионное оборудование,
- 63. Эффективное использование поверхности катализатора Улучшение пористой структуры – обеспечивающей повышение степени использования внутренней поверхности зерна (доступность
- 64. Зависимость поверхности контакта от геометрической формы и размера гранулы катализатора Лекции 1, 2
- 65. Размерный эффект в катализе. Эффект общей поверхности Увеличение концентрации граничных атомов Увеличение дефектности, появление новых дефектов
- 66. Лекции 1, 2 Методы приготовления катализаторов, основанные на механическом смешении компонентов подготовка исходных компонентов → смешение
- 67. Технология производства катализаторов методами смешения
- 68. Схема приготовления катализатора
- 69. Лекции 1, 2 механохимия В основу механохимических методов получения пористых материалов положены принципы физико-химической механики, состоящие
- 70. Механохимия Новым для технологии катализаторов и быстро развивающимся направлением модифицирования твердых тел является механические методы их
- 71. Механохимия При небольших статистических нагрузках вся механическая энергия переходит в тепловую. Более сильная статистическая или динамическая
- 72. механохимия Если механическое воздействие достаточно велико (значительно превышает предел прочности твердого тела) то происходит разрушение- диспергирование.
- 73. Время релаксации Время релаксации этого потенциала может длится секунды или годы. 1.Короткая время релаксации –тепловой энергии.
- 74. Лекции 1, 2 Метод термохимической активации кристаллических веществ Фазовые превращения кристаллических гидроксидов при их нагреве определяются
- 75. Лекции 1, 2 Термохимическая активация Таким образом, можно заключить, что термохимическая активация твердых веществ - процесс
- 76. Работа катализатора Согласно современным представлениям, катализатор образует комплекс с реагирующими молекулами, стабилизируемый химическими связями. После перегруппировки
- 77. Формирование рабочей поверхности катализатора Окончательные свойства катализаторов формируются под действием реакционной среды. Изменения состава катализаторов в
- 78. Механизм каталитических реакций Химическая реакция включает ряд элементарных процессов, которые могут протекать последовательно или параллельно. Эти
- 79. Элементарные процессы в гетерогенном катализе Не существует общепринятой классификации элементарных процессов гетерогенного катализа. *Адсорбция-десорбция включает процессы
- 80. Номенклатура поверхностных интермедиатов Лекции 1, 2
- 81. Номенклатура каталитических реакций В общем случае название каталитической реакции может образовываться добавлением прилагательного "каталитическая" к обычному
- 82. Предполагаемые стадии детального механизма реакции Лекции 1, 2 Поскольку скорость реакции в целом определяется скоростью наиболее
- 83. где [AS] - концентрация адсорбированной формы вещества А на поверхности катализатора, выражаемая обычно в долях единицы,
- 84. Вначале молекулы СО и Н2 адсорбируются на поверхности медного катализатора. Затем молекулы СО образуют с катализатором
- 85. В присутствии никелевого катализатора как СО, так и Н2 хемосорбируются на поверхности в диссоциированной форме, и
- 86. Работа катализатора Лекции 1, 2
- 87. Азот весьма инертное вещество. Для разрыва связи N-N в его молекуле необходима энергия порядка 200 ккал/моль.
- 88. Активность таких катализаторов, как алюмосиликаты, применяющихся при крекинге нефти, определяется присутствием на их поверхности кислот Бренстеда
- 89. Активность кислотных катализаторов обусловливается их способностью реагировать с углеводородами с образованием в качестве промежуточного продукта карбений-иона.
- 90. Дегидрирование парафинов на поверхности АЦ Лекции 1, 2
- 91. Причины дезактивации катализаторов
- 92. Общее изменение активности катализатора Изменения истинной и условной удельной активности, обусловленные изменением химического состава* Изменения наблюдаемой
- 93. 4 *Самахов А.А., Зайдман Н.М., Буянов Р.А. Об изменении активности катализаторов в процессе эксплуатации.- Новосибирск: Наука,
- 94. Изменения наблюдаемой активности, обусловленные изменением структуры (общей поверхности, поверхности активного компонента и степени их использования) Понижение
- 95. Время, в течение которого активность снижается до столь низкого уровня, что требуется замена катализатора или его
- 96. По литературным данным, полное подавление активности катализатора происходит при концентрации кокса на платине от 0,07-0.2 %масс.
- 97. Дезактивация металлических катализаторов Агрегация кристаллитов платины. Свежий катализатор содержат кристаллиты платины размером от 1 до 20
- 98. Соединения фосфора являются сильными ядами, их попадание возможно из добавок к смазочным маслам, используемых в насосах
- 99. Блокировка поверхности коксом Аморфный: С/Н =0,5-1,0; Т удаления500-550 С Графитизированный: С/Н =1,5-2,0;Т удаления 800-900С Пример: Механизм
- 100. Примеры дезактивации промышленных катализаторов Катализаторы оксихлорирования этилена дезактивируются за счет формирования низкоплавких комплексов меди. Изменение химического
- 101. Способы реактивации (регенерации) Способ низкотемпературного выжигания кокса с активной поверхности катализатора озоном, растворенным в сверхкритическом флюиде
- 102. Гидрирование этилена На пер в о м эт апе свобо д ны й прото н ст
- 104. Скачать презентацию

Катализаторы стратегическая продукция
Малым количеством катализатора можно превратить громадные количества вещества (фактор
Катализаторы стратегическая продукция
Малым количеством катализатора можно превратить громадные количества вещества (фактор
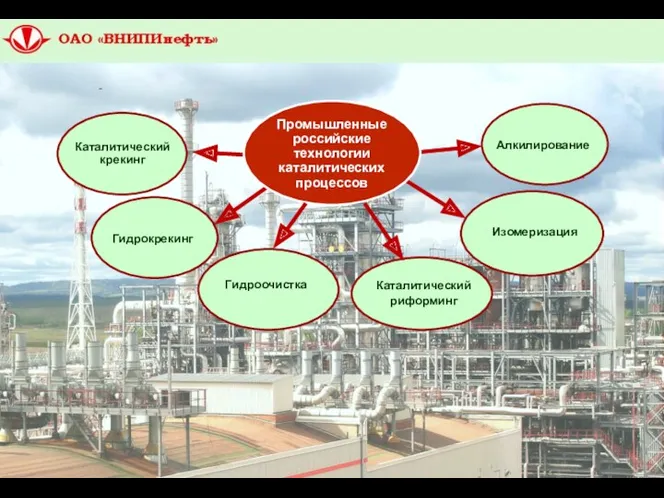
Промышленные
российские
технологии
каталитических
процессов
каталитических
процессов

Нефтехимия России
16
Нефтехимия России
16

Катализаторы стратегическая продукция
Объем мирового рынка катализаторов - 17,5 млрд.долл.США/год.
В России 15
Катализаторы стратегическая продукция
Объем мирового рынка катализаторов - 17,5 млрд.долл.США/год.
В России 15

Катализаторы стратегическая продукция
Зависимость отечественной технологии от поставок зарубежных катализаторов:
- Каталитический крекинг
Катализаторы стратегическая продукция
Зависимость отечественной технологии от поставок зарубежных катализаторов:
- Каталитический крекинг

Катализаторы стратегическая продукция
Анализ показывает масштабный спад в областях разработки, обновления ассортимента
Катализаторы стратегическая продукция
Анализ показывает масштабный спад в областях разработки, обновления ассортимента

Катализаторы стратегическая продукция
Причины
Наукоемкость ОТСТАВАНИЕ РОССИЙСКИХ РАЗРАБОТОК ВО МНОГИХ ОБЛАСТЯХ КАТАЛИТИЧЕСКИХ ТЕХНОЛОГИЙ
Плохая
Катализаторы стратегическая продукция
Причины
Наукоемкость ОТСТАВАНИЕ РОССИЙСКИХ РАЗРАБОТОК ВО МНОГИХ ОБЛАСТЯХ КАТАЛИТИЧЕСКИХ ТЕХНОЛОГИЙ
Плохая

Катализаторы стратегическая продукция
Пути решения
Создание условий для приоритетного использования отечественных НТР
Катализаторы стратегическая продукция
Пути решения
Создание условий для приоритетного использования отечественных НТР
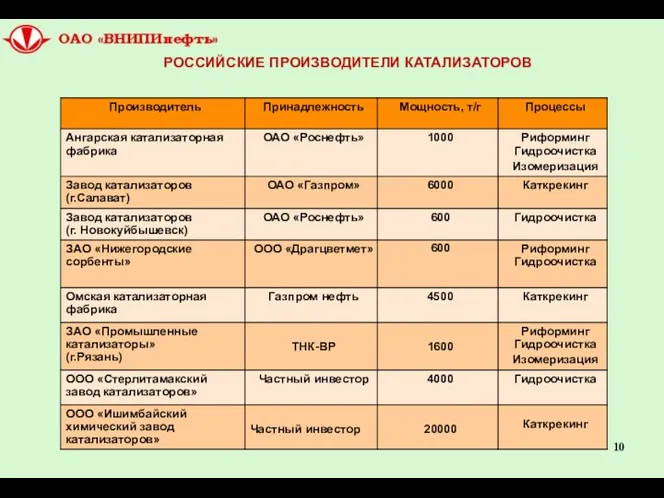
РОССИЙСКИЕ ПРОИЗВОДИТЕЛИ КАТАЛИЗАТОРОВ
РОССИЙСКИЕ ПРОИЗВОДИТЕЛИ КАТАЛИЗАТОРОВ

Прогнозируемая годовая потребность России в катализаторах гидроочистки
Современные промышленные катализаторы гидроочистки
Нанесённые зарубежные
Прогнозируемая годовая потребность России в катализаторах гидроочистки
Современные промышленные катализаторы гидроочистки
Нанесённые зарубежные
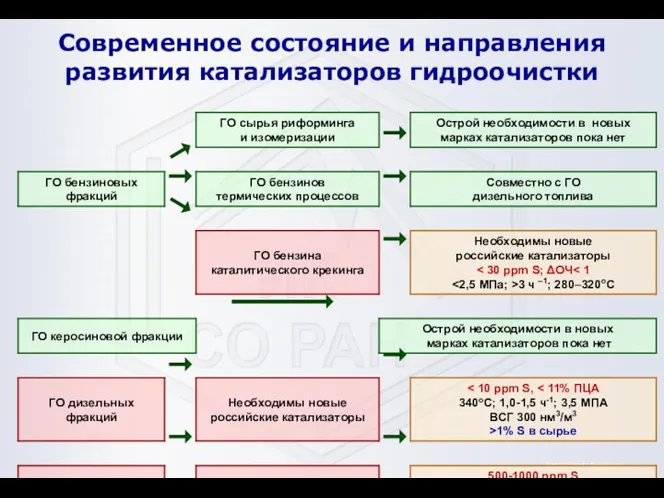
Современное состояние и направления развития катализаторов гидроочистки
Современное состояние и направления развития катализаторов гидроочистки

Лекции 1, 2
Исторические корни катализа
XV век: алхимики
1669 г.: Бекер
1759 г.: Шееле,
Лекции 1, 2
Исторические корни катализа
XV век: алхимики
1669 г.: Бекер
1759 г.: Шееле,

Лекции 1, 2
Исторические корни катализа
1806 г.: Клеман, Дезорм
1811 г.: Кирхгоф
2SO2 +
Лекции 1, 2
Исторические корни катализа
1806 г.: Клеман, Дезорм
1811 г.: Кирхгоф
2SO2 +

Лекции 1, 2
Исторические корни катализа
1813 г.: Тенар
1817 г.: Дэви
1818 г.:
Лекции 1, 2
Исторические корни катализа
1813 г.: Тенар
1817 г.: Дэви
1818 г.:

Лекции 1, 2
Становление катализа
1834 г.: Митчерлих
ввел понятие «контактные реакции»
1835 г.:
Лекции 1, 2
Становление катализа
1834 г.: Митчерлих
ввел понятие «контактные реакции»
1835 г.:

Лекции 1, 2
Основные положения определения катализа
À Возбуждение химических реакций (а не
Лекции 1, 2
Основные положения определения катализа
À Возбуждение химических реакций (а не

Лекции 1, 2
Основные положения определения катализа
Лекции 1, 2
Основные положения определения катализа

Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1875 г.:
Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1875 г.:

Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1930-1932 г.г.:
Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1930-1932 г.г.:

Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1950-1960 г.г.:
Лекции 1, 2
Роль катализа в становлении и развитии современной промышленности
1950-1960 г.г.:
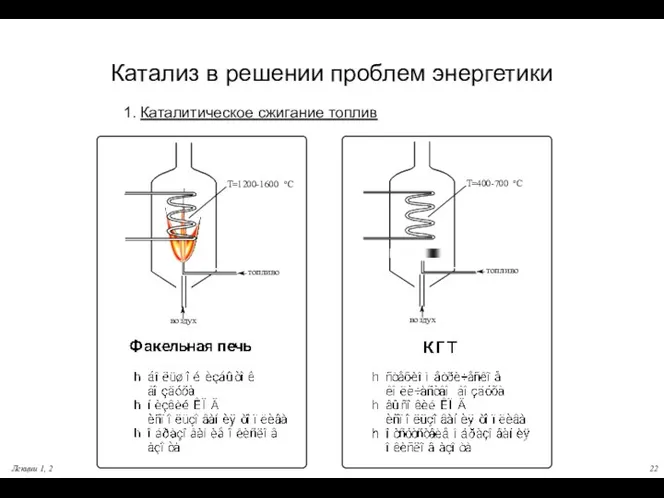
Лекции 1, 2
Катализ в решении проблем энергетики
1. Каталитическое сжигание топлив
Лекции 1, 2
Катализ в решении проблем энергетики
1. Каталитическое сжигание топлив
Лекции 1, 2
Катализ в решении проблем энергетики
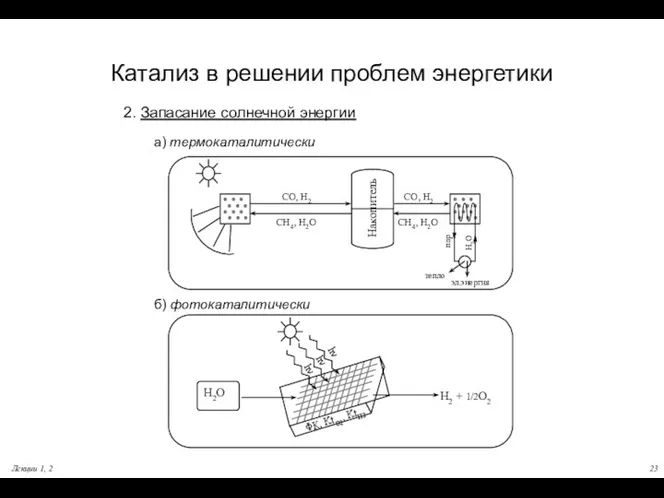
2. Запасание солнечной энергии
а) термокаталитически
б)
Лекции 1, 2
Катализ в решении проблем энергетики
2. Запасание солнечной энергии
а) термокаталитически
б)
Лекции 1, 2
Катализ в решении проблем энергетики
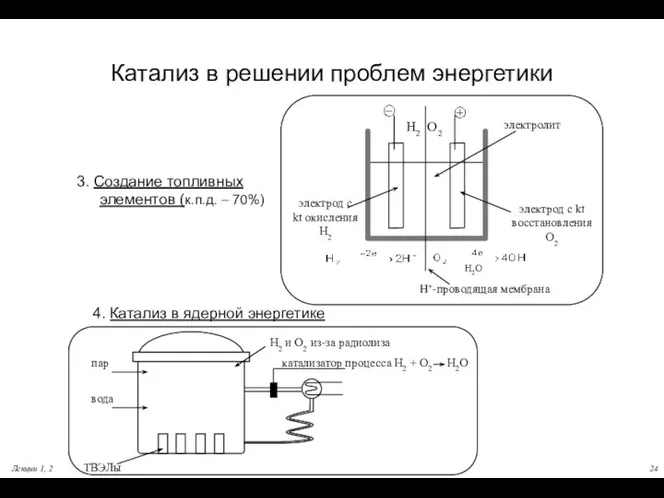
3. Создание топливных
элементов (к.п.д.
Лекции 1, 2
Катализ в решении проблем энергетики
3. Создание топливных элементов (к.п.д.
Лекции 1, 2
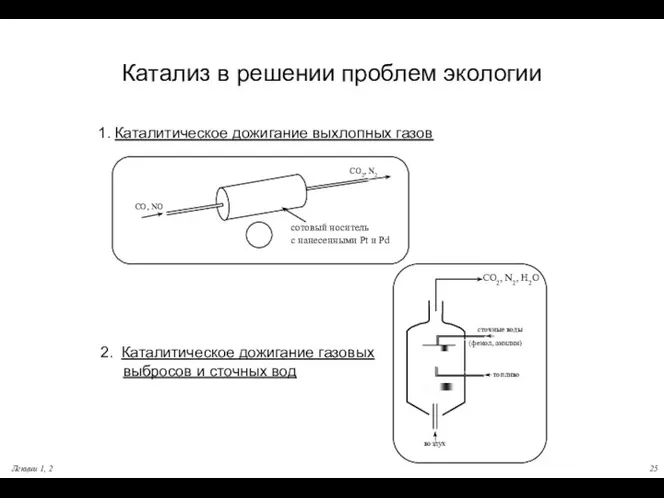
Катализ в решении проблем экологии
топливо
воздух
сточные воды
(фенол, анилин)
CO2, N2,
Лекции 1, 2
Катализ в решении проблем экологии
топливо
воздух
сточные воды
(фенол, анилин)
CO2, N2,

Лекции 1, 2
Катализ в живой природе
Почти все реакции в клетках
Лекции 1, 2
Катализ в живой природе
Почти все реакции в клетках

Лекции 1, 2
Катализ в живой природе
И.П.Павлов назвал ферменты “возбудителями жизни“
Число найденных
Лекции 1, 2
Катализ в живой природе
И.П.Павлов назвал ферменты “возбудителями жизни“
Число найденных

Лекции 1, 2
Классификация катализаторов
À Катализатор может быть как индивидуальным веществом, так
Лекции 1, 2
Классификация катализаторов
À Катализатор может быть как индивидуальным веществом, так
Лекции 1, 2
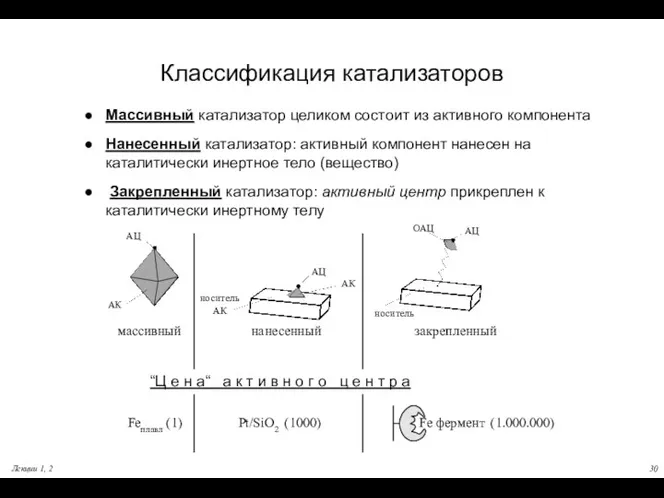
Классификация катализаторов
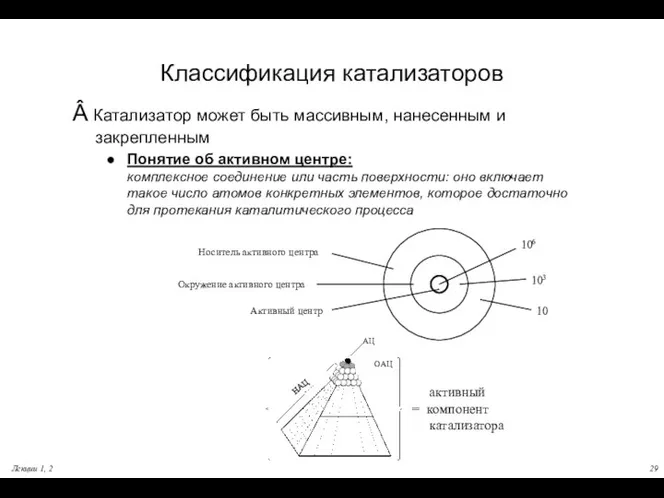
 Катализатор может быть массивным, нанесенным и закрепленным
Понятие
Лекции 1, 2
Классификация катализаторов
 Катализатор может быть массивным, нанесенным и закрепленным
Понятие
Лекции 1, 2
Классификация катализаторов
Массивный катализатор целиком состоит из активного компонента
Нанесенный катализатор:
Лекции 1, 2
Классификация катализаторов
Массивный катализатор целиком состоит из активного компонента
Нанесенный катализатор:

Лекции 1, 2
Классификация каталитических процессов
À Гомогенный катализ
 Ферментативный
катализ
Лекции 1, 2
Классификация каталитических процессов
À Гомогенный катализ
 Ферментативный
катализ
Лекции 1, 2
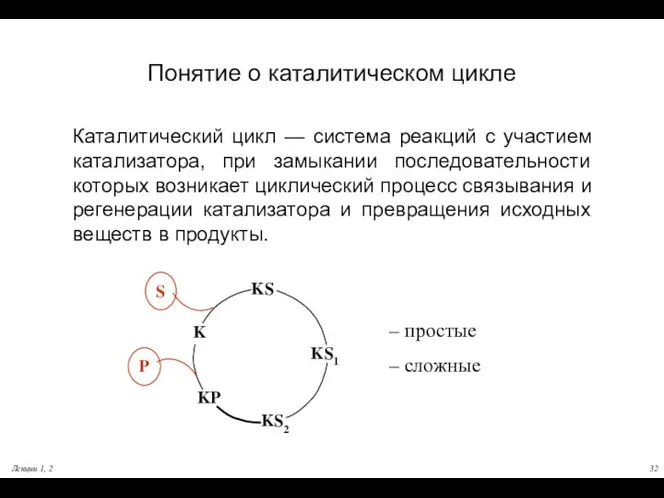
Понятие о каталитическом цикле
Каталитический цикл — система реакций с
Лекции 1, 2
Понятие о каталитическом цикле
Каталитический цикл — система реакций с

Лекции 1, 2
Каталитическая активность
число оборотов
Лекции 1, 2
Каталитическая активность
число оборотов
Лекции 1, 2
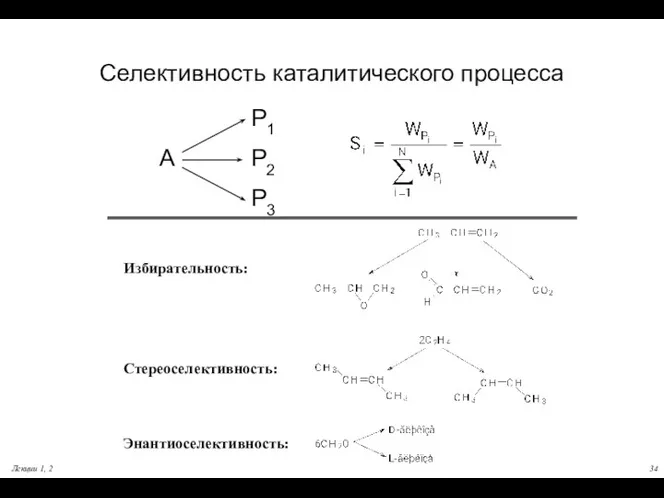
Селективность каталитического процесса
Лекции 1, 2
Селективность каталитического процесса
Лекции 1, 2
Лекции 1, 2

Лекции 1, 2
Для рентабельной эксплуатации катализатор должен обладать
1.Высокой каталитической активностью и
Лекции 1, 2
Для рентабельной эксплуатации катализатор должен обладать
1.Высокой каталитической активностью и

Лекции 1, 2
Стабильность
К числу главных характеристик катализатора относится его устойчивость
Лекции 1, 2
Стабильность
К числу главных характеристик катализатора относится его устойчивость

Лекции 1, 2
дисперсность
Термин дисперсность – производный от лат. слова dispersus (рассеянный,
Лекции 1, 2
дисперсность
Термин дисперсность – производный от лат. слова dispersus (рассеянный,

Лекции 1, 2
Механическая прочность
Прочность тонкодисперсного твердого тела в значительной степени зависит
Лекции 1, 2
Механическая прочность
Прочность тонкодисперсного твердого тела в значительной степени зависит

Лекции 1, 2
Определение и характеристики пористых материалов.
Мы знаем, что существуют понятия
Лекции 1, 2
Определение и характеристики пористых материалов.
Мы знаем, что существуют понятия

Лекции 1, 2
Супрамолекулярная структура пористого материала.
Пористые материалы характеризуются величинами удельной поверхности,
Лекции 1, 2
Супрамолекулярная структура пористого материала.
Пористые материалы характеризуются величинами удельной поверхности,

Лекции 1, 2
классификации пористых тел по размерам пор
Действительно, каково бы
Лекции 1, 2
классификации пористых тел по размерам пор
Действительно, каково бы
Лекции 1, 2
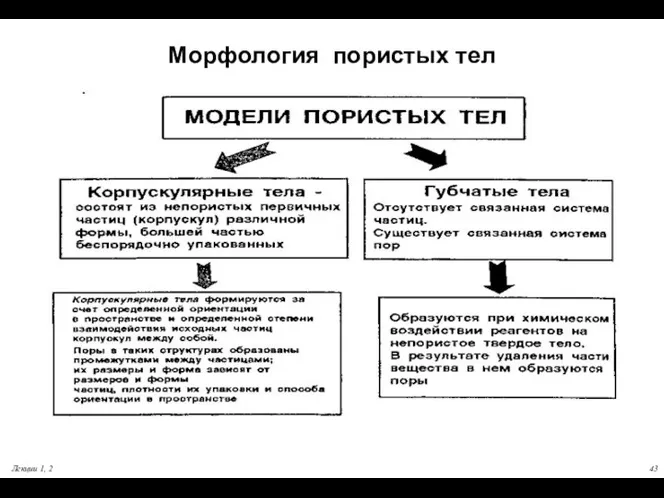
Морфология пористых тел
Лекции 1, 2
Морфология пористых тел

Лекции 1, 2
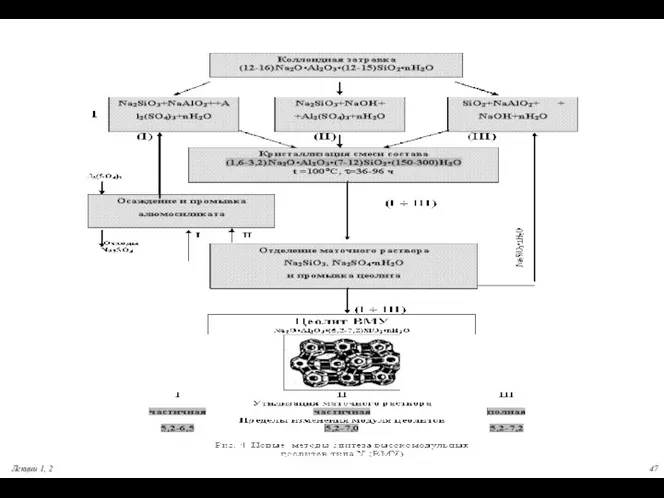
СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ
Известны цеолиты различных типов. Тип, к которому относится
Лекции 1, 2
СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ
Известны цеолиты различных типов. Тип, к которому относится
![Лекции 1, 2 СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ [Мn]+ [(Al2O3)m×(SiO2)k×(H2O)l]- [Мn]+= H+, K+](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/324473/slide-44.jpg)
Лекции 1, 2
СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ
[Мn]+ [(Al2O3)m×(SiO2)k×(H2O)l]-
[Мn]+= H+, K+ , Na+, Ca2+….
Лекции 1, 2
СИНТЕТИЧЕСКИЕ ЦЕОЛИТЫ
[Мn]+ [(Al2O3)m×(SiO2)k×(H2O)l]-
[Мn]+= H+, K+ , Na+, Ca2+….

Объемы производства
Мировое –1,5 млн. тонн в год (~5 млрд. $);
Россия
Объемы производства Мировое –1,5 млн. тонн в год (~5 млрд. $); Россия
Лекции 1, 2
Лекции 1, 2
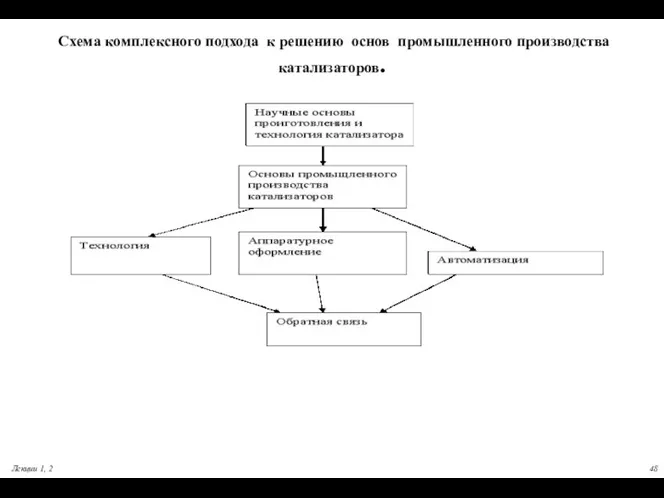
Схема комплексного подхода к решению основ промышленного производства катализаторов.
Лекции 1,
Схема комплексного подхода к решению основ промышленного производства катализаторов.
Лекции 1,

Лекции 1, 2
Традиционные методы синтеза пористых материалов - катализаторов
1. Осаждение (соосаждение
Лекции 1, 2
Традиционные методы синтеза пористых материалов - катализаторов
1. Осаждение (соосаждение
Лекции 1, 2
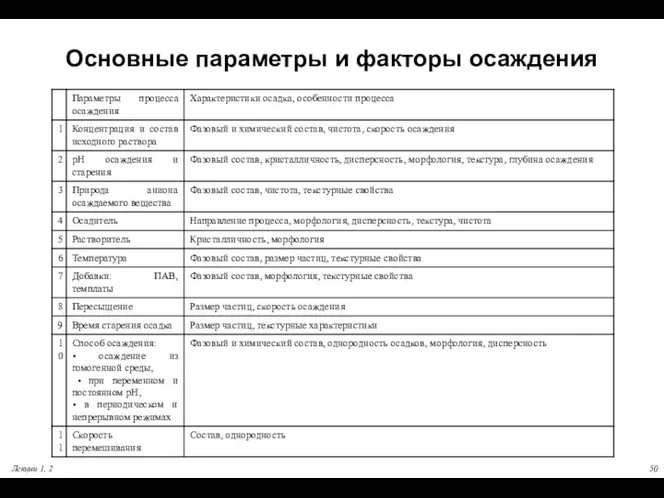
Основные параметры и факторы осаждения
Лекции 1, 2
Основные параметры и факторы осаждения
Лекции 1, 2
.
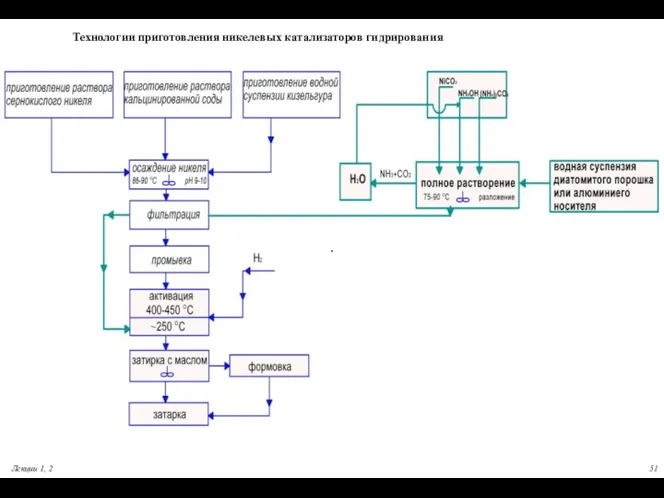
Технологии приготовления никелевых катализаторов гидрирования
Лекции 1, 2
.
Технологии приготовления никелевых катализаторов гидрирования


Восстановительное разложение основного карбоната никеля (активация)
Восстановительное разложение основного карбоната никеля (активация)
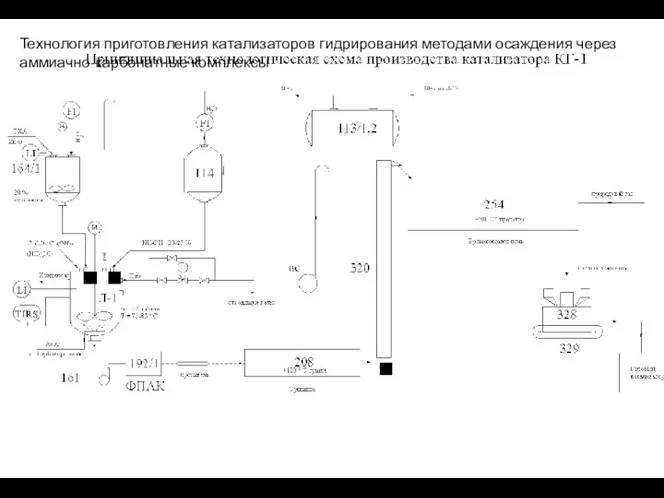
Технология приготовления катализаторов гидрирования методами осаждения через аммиачно-карбонатные комплексы
Технология приготовления катализаторов гидрирования методами осаждения через аммиачно-карбонатные комплексы

Технология приготовления.
Алюмо-цинк-медные катализаторы гидрирования
1. Приготовление аммиачно-карбонатного комплекса меди
Карбонатный медно-аммиачный
Технология приготовления.
Алюмо-цинк-медные катализаторы гидрирования
1. Приготовление аммиачно-карбонатного комплекса меди
Карбонатный медно-аммиачный

Лекции 1, 2
Физико-химические основы получения пористых материалов методами нанесения
Требования, предъявляемые к
Лекции 1, 2
Физико-химические основы получения пористых материалов методами нанесения
Требования, предъявляемые к

Лекции 1, 2
Способы нанесения
Состояние и дисперсность активного компонента в нанесенных катализаторах
Лекции 1, 2
Способы нанесения
Состояние и дисперсность активного компонента в нанесенных катализаторах

Лекции 1, 2
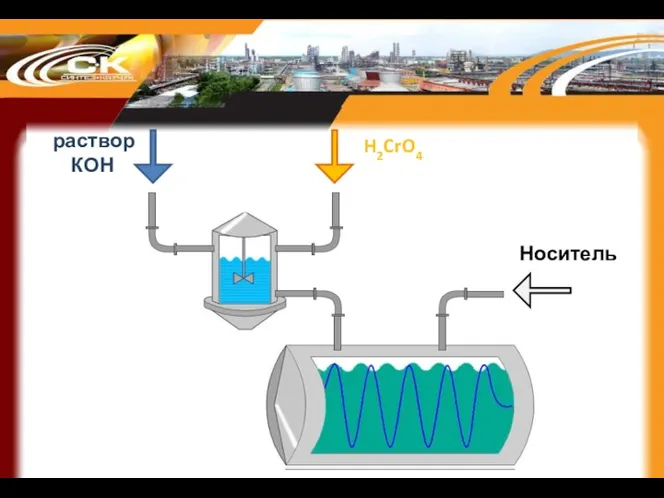
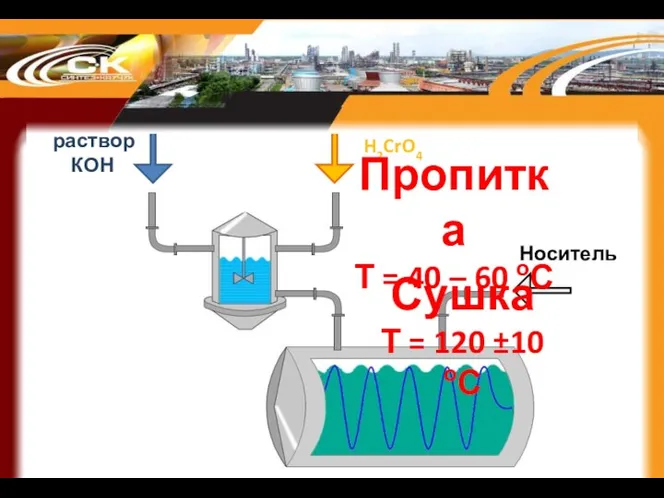
Режимы пропитки
Капиллярная пропитка. Нанесение вещества осуществляется за счет
Лекции 1, 2
Режимы пропитки
Капиллярная пропитка. Нанесение вещества осуществляется за счет
раствор
КОН
H2CrO4
Носитель
раствор
КОН
H2CrO4
Носитель
раствор
КОН
H2CrO4
Носитель
Сушка
Т = 120 ±10 оС
Пропитка
Т = 40 –
раствор
КОН
H2CrO4
Носитель
Сушка
Т = 120 ±10 оС
Пропитка
Т = 40 –
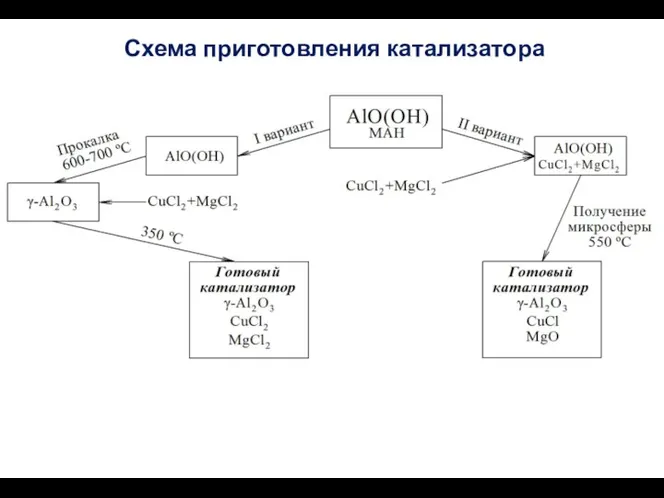
Схема приготовления катализатора
Схема приготовления катализатора

Способы формовки
Таблетирование (штамбовка)
формование порошков методом таблетирования
Преимущество: строго определенный длины
Недостатки:
Способы формовки
Таблетирование (штамбовка)
формование порошков методом таблетирования
Преимущество: строго определенный длины
Недостатки:

Эффективное использование поверхности катализатора
Улучшение пористой структуры – обеспечивающей повышение степени использования
Эффективное использование поверхности катализатора
Улучшение пористой структуры – обеспечивающей повышение степени использования

Зависимость поверхности контакта от геометрической формы и размера гранулы катализатора
Лекции 1,
Зависимость поверхности контакта от геометрической формы и размера гранулы катализатора
Лекции 1,

Размерный эффект в катализе.
Эффект общей поверхности
Увеличение концентрации граничных атомов
Увеличение дефектности, появление
Размерный эффект в катализе.
Эффект общей поверхности
Увеличение концентрации граничных атомов
Увеличение дефектности, появление

Лекции 1, 2
Методы приготовления катализаторов, основанные
на механическом смешении компонентов
подготовка исходных
Лекции 1, 2
Методы приготовления катализаторов, основанные
на механическом смешении компонентов
подготовка исходных
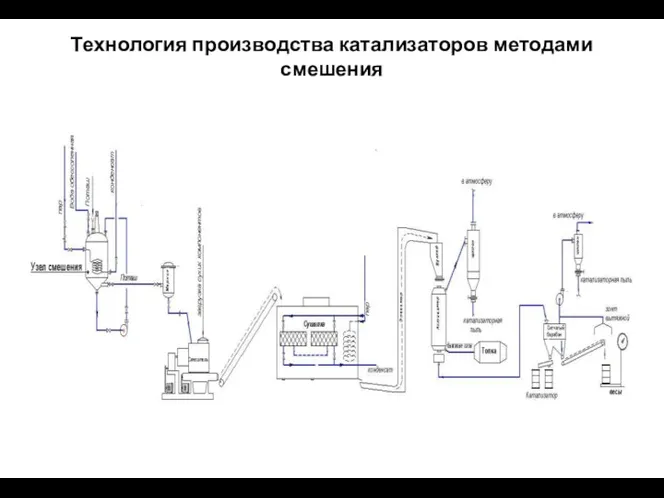
Технология производства катализаторов методами смешения
Технология производства катализаторов методами смешения
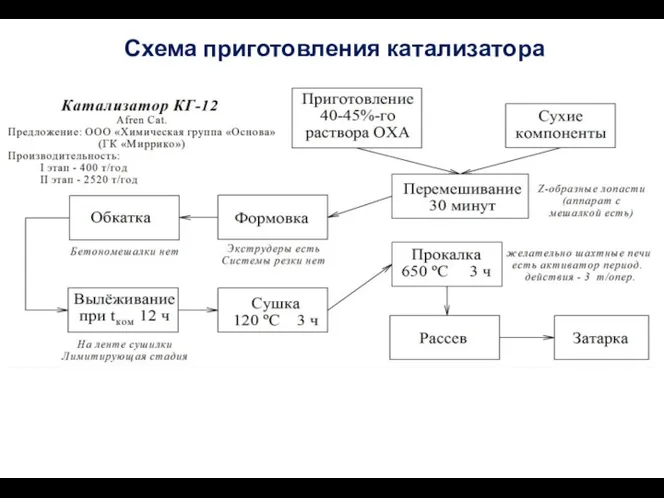
Схема приготовления катализатора
Схема приготовления катализатора

Лекции 1, 2
механохимия
В основу механохимических методов получения пористых материалов положены
Лекции 1, 2
механохимия
В основу механохимических методов получения пористых материалов положены

Механохимия
Новым для технологии катализаторов и быстро развивающимся направлением модифицирования твердых тел
Механохимия
Новым для технологии катализаторов и быстро развивающимся направлением модифицирования твердых тел

Механохимия
При небольших статистических нагрузках вся механическая энергия переходит в тепловую.
Более
Механохимия
При небольших статистических нагрузках вся механическая энергия переходит в тепловую.
Более

механохимия
Если механическое воздействие достаточно велико (значительно превышает предел прочности твердого тела)
механохимия
Если механическое воздействие достаточно велико (значительно превышает предел прочности твердого тела)

Время релаксации
Время релаксации этого потенциала может длится секунды или годы.
1.Короткая время
Время релаксации
Время релаксации этого потенциала может длится секунды или годы.
1.Короткая время

Лекции 1, 2
Метод термохимической активации кристаллических веществ
Фазовые превращения кристаллических гидроксидов при
Лекции 1, 2
Метод термохимической активации кристаллических веществ
Фазовые превращения кристаллических гидроксидов при

Лекции 1, 2
Термохимическая активация
Таким образом, можно заключить, что термохимическая активация твердых
Лекции 1, 2
Термохимическая активация
Таким образом, можно заключить, что термохимическая активация твердых

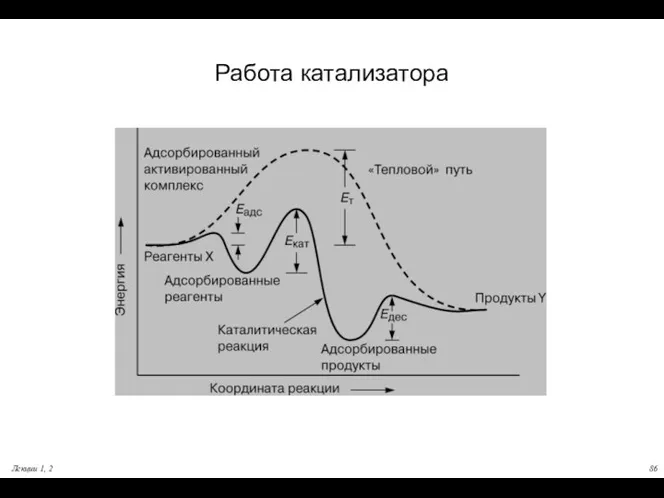
Работа катализатора
Согласно современным представлениям, катализатор образует комплекс с реагирующими молекулами, стабилизируемый
Работа катализатора
Согласно современным представлениям, катализатор образует комплекс с реагирующими молекулами, стабилизируемый

Формирование рабочей поверхности катализатора
Окончательные свойства катализаторов формируются под действием реакционной среды.
Формирование рабочей поверхности катализатора
Окончательные свойства катализаторов формируются под действием реакционной среды.

Механизм каталитических реакций
Химическая реакция включает ряд элементарных процессов, которые могут протекать
Механизм каталитических реакций
Химическая реакция включает ряд элементарных процессов, которые могут протекать

Элементарные процессы в гетерогенном катализе
Не существует общепринятой классификации элементарных процессов гетерогенного
Элементарные процессы в гетерогенном катализе Не существует общепринятой классификации элементарных процессов гетерогенного

Номенклатура поверхностных интермедиатов
Лекции 1, 2
Номенклатура поверхностных интермедиатов
Лекции 1, 2

Номенклатура каталитических реакций
В общем случае название каталитической реакции может образовываться добавлением
Номенклатура каталитических реакций
В общем случае название каталитической реакции может образовываться добавлением

Предполагаемые стадии детального механизма реакции
Лекции 1, 2
Поскольку скорость реакции в
Предполагаемые стадии детального механизма реакции
Лекции 1, 2
Поскольку скорость реакции в
![где [AS] - концентрация адсорбированной формы вещества А на поверхности](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/324473/slide-82.jpg)
где [AS] - концентрация адсорбированной формы вещества А на поверхности
где [AS] - концентрация адсорбированной формы вещества А на поверхности
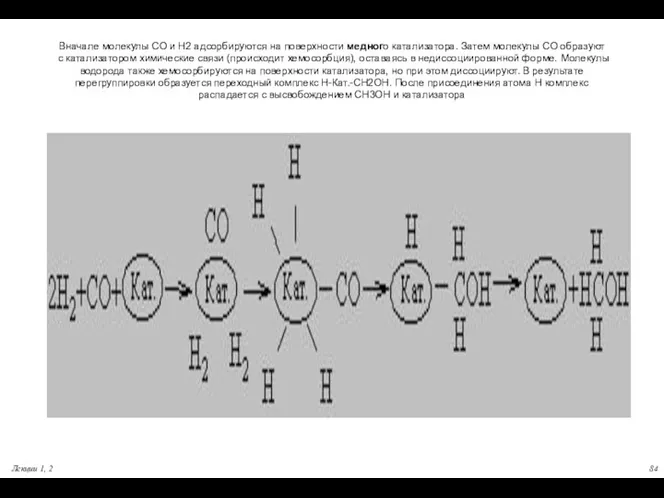
Вначале молекулы СО и Н2 адсорбируются на поверхности медного катализатора. Затем
Вначале молекулы СО и Н2 адсорбируются на поверхности медного катализатора. Затем
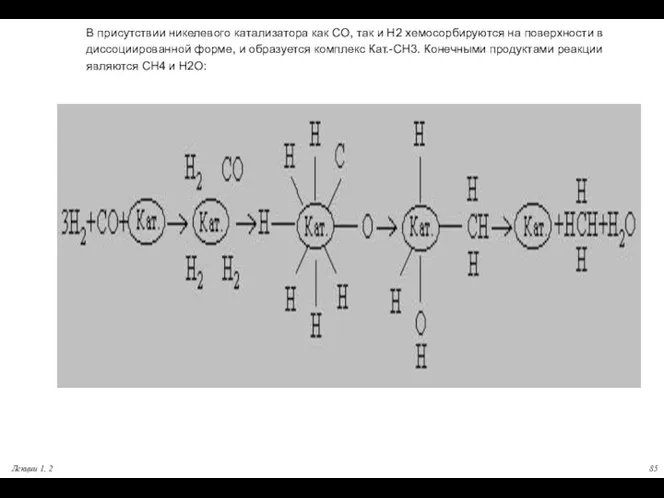
В присутствии никелевого катализатора как СО, так и Н2 хемосорбируются на
В присутствии никелевого катализатора как СО, так и Н2 хемосорбируются на
Работа катализатора
Лекции 1, 2
Работа катализатора
Лекции 1, 2

Азот весьма инертное вещество. Для разрыва связи N-N в его
Азот весьма инертное вещество. Для разрыва связи N-N в его

Активность таких катализаторов, как алюмосиликаты, применяющихся при крекинге нефти, определяется присутствием
Активность таких катализаторов, как алюмосиликаты, применяющихся при крекинге нефти, определяется присутствием
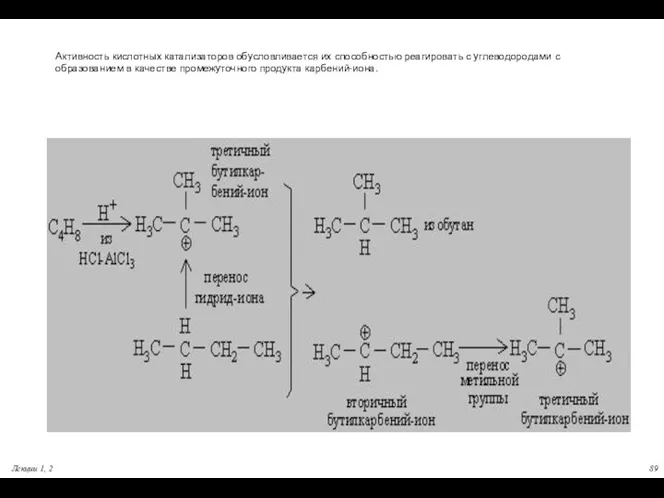
Активность кислотных катализаторов обусловливается их способностью реагировать с углеводородами с образованием
Активность кислотных катализаторов обусловливается их способностью реагировать с углеводородами с образованием
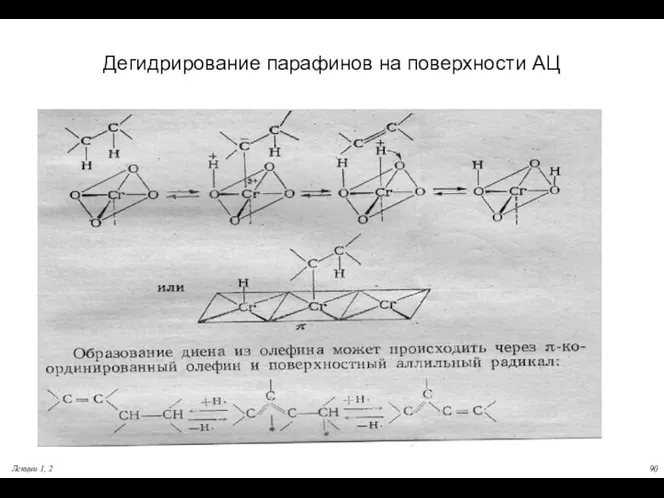
Дегидрирование парафинов на поверхности АЦ
Лекции 1, 2
Дегидрирование парафинов на поверхности АЦ
Лекции 1, 2
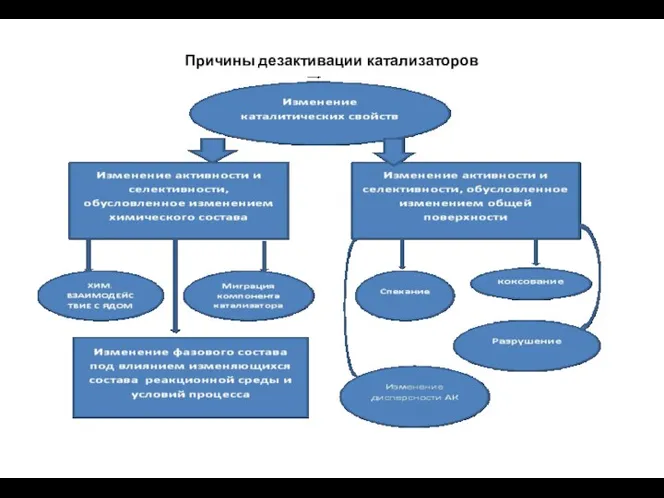
Причины дезактивации катализаторов
Причины дезактивации катализаторов
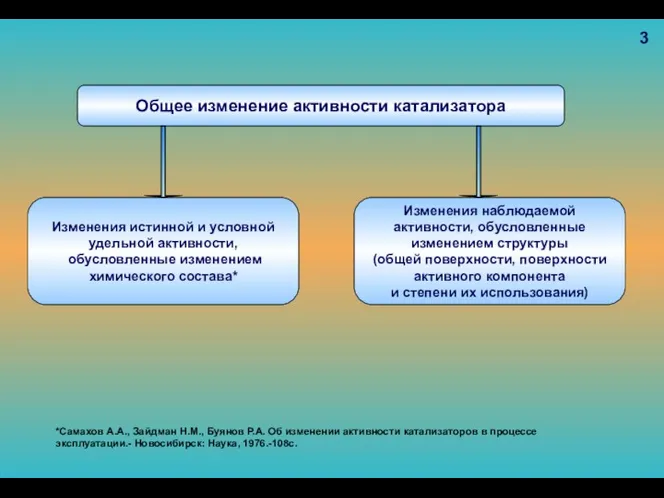
Общее изменение активности катализатора
Изменения истинной и условной
удельной активности,
обусловленные изменением
Общее изменение активности катализатора
Изменения истинной и условной
удельной активности,
обусловленные изменением
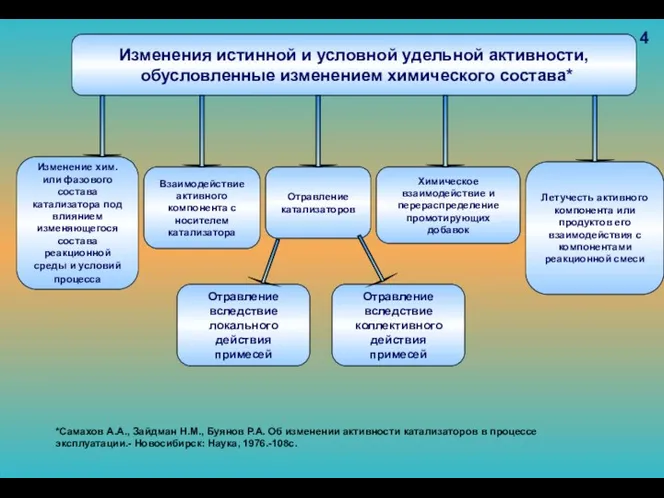
4
*Самахов А.А., Зайдман Н.М., Буянов Р.А. Об изменении активности катализаторов в
4
*Самахов А.А., Зайдман Н.М., Буянов Р.А. Об изменении активности катализаторов в
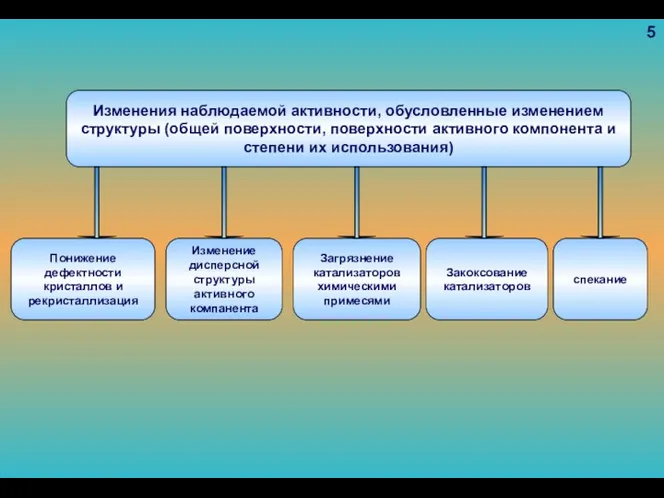
Изменения наблюдаемой активности, обусловленные изменением структуры (общей поверхности, поверхности активного компонента
Изменения наблюдаемой активности, обусловленные изменением структуры (общей поверхности, поверхности активного компонента

Время, в течение которого активность снижается до столь низкого уровня, что
Время, в течение которого активность снижается до столь низкого уровня, что
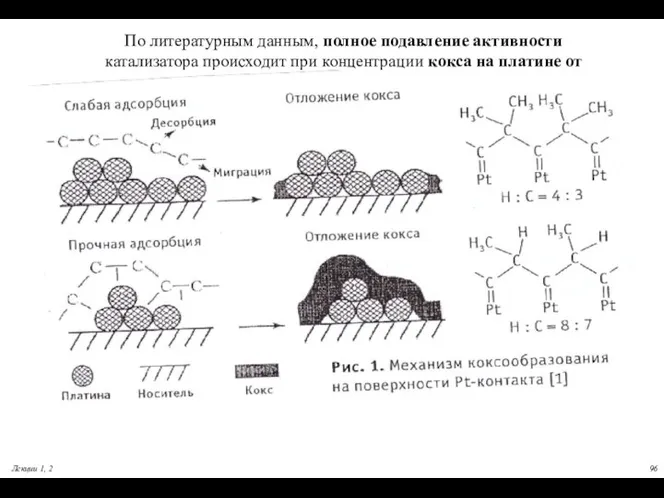
По литературным данным, полное подавление активности катализатора происходит при концентрации кокса
По литературным данным, полное подавление активности катализатора происходит при концентрации кокса

Дезактивация металлических катализаторов
Агрегация кристаллитов платины. Свежий катализатор содержат кристаллиты платины
Дезактивация металлических катализаторов
Агрегация кристаллитов платины. Свежий катализатор содержат кристаллиты платины

Соединения фосфора являются сильными ядами, их попадание возможно из добавок к
Соединения фосфора являются сильными ядами, их попадание возможно из добавок к

Блокировка поверхности коксом
Аморфный: С/Н =0,5-1,0; Т удаления500-550 С
Графитизированный: С/Н =1,5-2,0;Т удаления
Блокировка поверхности коксом
Аморфный: С/Н =0,5-1,0; Т удаления500-550 С
Графитизированный: С/Н =1,5-2,0;Т удаления

Примеры дезактивации промышленных катализаторов
Катализаторы оксихлорирования этилена дезактивируются за счет формирования низкоплавких
Примеры дезактивации промышленных катализаторов
Катализаторы оксихлорирования этилена дезактивируются за счет формирования низкоплавких

Способы реактивации (регенерации)
Способ низкотемпературного выжигания кокса с активной поверхности катализатора озоном,
Способы реактивации (регенерации)
Способ низкотемпературного выжигания кокса с активной поверхности катализатора озоном,
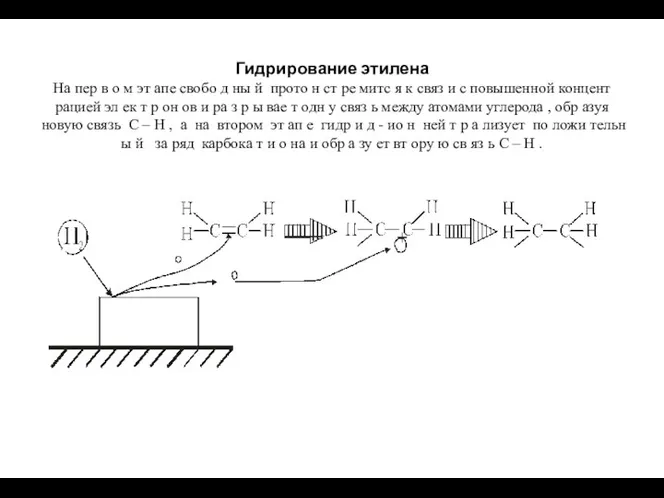
Гидрирование этилена
На пер в о м эт апе свобо д ны
Гидрирование этилена На пер в о м эт апе свобо д ны
 Аминокислоты, их строение, изомерия, свойства, применение
Аминокислоты, их строение, изомерия, свойства, применение Изучение процесса коррозии железа (домашний эксперимент)
Изучение процесса коррозии железа (домашний эксперимент) Минералы и горные породы
Минералы и горные породы Сұйықтардағы газ ерітінділері. Генри заңы. Сұйық-сұйық ерітінділердегі бу қысымы. Рауль заңынын ауытқу. Криометрия
Сұйықтардағы газ ерітінділері. Генри заңы. Сұйық-сұйық ерітінділердегі бу қысымы. Рауль заңынын ауытқу. Криометрия Протекание ОВР зависит от рН среды
Протекание ОВР зависит от рН среды Сполуки неметалічних елементів з Гідрогеном
Сполуки неметалічних елементів з Гідрогеном Хром. Химиялық қасиеттері
Хром. Химиялық қасиеттері 20230306_vodorod_ego_fizicheskie_svoystva
20230306_vodorod_ego_fizicheskie_svoystva Закономерности процессов нитрования НЦ
Закономерности процессов нитрования НЦ Основные электрохимические процессы
Основные электрохимические процессы Иодометрия. Комплексиметрия (начало)
Иодометрия. Комплексиметрия (начало) Золото Au (Аурум)
Золото Au (Аурум) Кислородсодержащие органические соединения. Спирты
Кислородсодержащие органические соединения. Спирты Растворение как физико-химический процесс
Растворение как физико-химический процесс Химический анализ веществ
Химический анализ веществ Диагностические свойства минералов. Занятие 3-4
Диагностические свойства минералов. Занятие 3-4 Химия воды
Химия воды Химические свойства основных неорганических соединений в свете ЭД и ОВР
Химические свойства основных неорганических соединений в свете ЭД и ОВР Основные виды химических связей
Основные виды химических связей Занимательные опыты
Занимательные опыты Титриметрический метод анализа
Титриметрический метод анализа Введение в органическую химию
Введение в органическую химию Полиэтилен - термопластичный полимер этилена
Полиэтилен - термопластичный полимер этилена Native elements
Native elements Понятие о солях (8 класс)
Понятие о солях (8 класс) Скорость химической реакции
Скорость химической реакции Состояние радионуклидов в различных фазах и методы его изучения
Состояние радионуклидов в различных фазах и методы его изучения Периодический закон и периодический закон Д.И. Менделеева
Периодический закон и периодический закон Д.И. Менделеева