- Методы решения электронного уравнения Шредингера

Содержание
- 2. В расчетных методах квантовой химии широко применяется приближение Борна-Оппенгеймера (лекция № 3). Оно позволяет заменить стационарное
- 3. Решение электронного УШ затруднено наличием оператора энергии взаимодействия электронов: здесь rij – расстояние между электронами i
- 4. Используя (4.2), Хартри заменил оператор (4.1) на эффективный электронный потенциал, описывающий взаимодействие электрона i с другими
- 5. Для того, чтобы провести усреднение , надо уже знать функции, описывающие электроны j ≠ i! Как
- 6. и строят оператор (h0i)ССП. Затем решают набор одно-электронных уравнений Хартри (4.3). Полученные решения χ1j(rj) используют, чтобы
- 7. Многоэлектронная волновая функция ϕ(r) системы с замкнутой оболочкой, может быть описана единственным детерминантом Слэйтера (лекция 3).
- 8. Метод Хартри-Фока Fφi = εi φi Как выглядят орбитали? Аппроксимация многоэлектронной волновой функция единственным детерминантом Слэйтера
- 9. Приближение МО ЛКАО Молекулярная Орбиталь – Линейная Комбинация Атомных Орбиталей: M атомных орбиталей (АО) χμ называются
- 10. Выбор базисных АО Должны давать хорошее приближение к истинной ВФ (например, возле ядер и на больших
- 11. АО состоит из угловой и радиальной частей. В качестве угловой части используются сферические гармоники Ylm. Поэтому
- 12. OCT (STO) Функции STO – точные решения для радиальных частей орбиталей водородоподобного атома. Имеют «правильное» поведение
- 13. ОГТ (GTO) Допускают быстрое вычисление интегралов (более удобны в вычислениях). Не имеют физического смысла. Неправильные асимптотики
- 14. Общее резюме Более физичные и «правильные» STO мало пригодны для вычислений. Нефизичные с неправильной асимптотикой GTO
- 15. Метод Хартри-Фока-Рутана. Базисные функции, обычно центрированы на атомах (Атомные Орбитали). АО «образуют» базис для представления МО.
- 16. Уравнения Хартри-Фока. Подставим в них разложение МО по АО. В обозначениях Дирака умножим на χν* и
- 17. Обозначим Sνμ – матрица интегралов перекрывания, её элементы говорят о степени пространственного перекрывания АО χν и
- 18. Матричная форма уравнений Хартри-Фока-Рутана F – матрица Фока с элементами Fνμ, S – матрица перекрывания с
- 19. При решении уравнений Хартри-Фока-Рутана, кроме интеграла перекрывания, необходимо вычислять большое число одноэлектронных hμν = = ∫χμ(1)hχν(1)dτ1
- 20. Методы решения электронного уравнения Шредингера. Неэмпирические (ab initio) методы расчета (лабораторная работа № 1) Полуэмпирические методы
- 21. Основная проблема – расчет интегралов на АО Молекула СО – самый простой базисный набор (минимальный базис):
- 22. Неэмпирические методы расчета: Орбитали слейтеровского типа (STO) обычно аппроксимируются орбиталями гауссова типа (ОГТ). Это обусловливает использование
- 23. Типы базисных наборов (по числу базисных функций) Минимальный базисный набор – используется только одна функция на
- 24. Расширенный базисный набор Используя две, три и т.д. функций на одну орбиталь: можно добиться лучшего описания
- 25. Точность ab initio методов Ошибки ab initio расчета в основном обусловлены недостаточно полным учетом электронной корреляции
- 26. Полуэмпирические методы квантовой химии Рассматриваем методы на основе приближения ХФ. В методе ХФ основные затраты —
- 27. Общие требования к полуэмпирическим методам Хотим… Высокая скорость расчета БОЛЬШИХ систем. Надежные результаты. Легко интерпретируемые результаты.
- 28. Общие приближения полуэмпирических методов Валентное приближение. Явно рассматриваются только валентные электроны Минимальный базисный набор ортогональных АО.
- 29. Приближение НДП АО экспоненциально затухают с удалением от ядра. Если орбитали центрированы на разных атомах A
- 30. Если АО принадлежат одному атому, то они ортогональны и 3) Только для одинаковых орбиталей одного и
- 31. В результате применения приближения НДП Все трех- и четырехцентровые кулоновские и все обменные интегралы исчезают: 4-хмерный
- 32. Плата за резкое упрощение. Появляется большое количество параметров, численные значения которых находят из эксперимента или неэмпирического
- 33. Метод MNDO (МПДП) Modified Neglect of Diatomic Overlap (Модифицированное Пренебрежение Двухатомным Перекрыванием) В отличие от методов
- 34. Методы AM1 и PM3 Специально параметризованы для описания водородных связей. АМ1 (Austin Model 1) – разработана
- 35. Полуэмпирические методы, учитывающие электронную корреляцию, CNDO/S и INDO/S Они параметризованы для расчетов электронные спектров органических молекул.
- 37. Скачать презентацию

В расчетных методах квантовой химии широко применяется приближение Борна-Оппенгеймера (лекция №
В расчетных методах квантовой химии широко применяется приближение Борна-Оппенгеймера (лекция №

Решение электронного УШ затруднено наличием
оператора энергии взаимодействия электронов:
здесь rij – расстояние
Решение электронного УШ затруднено наличием
оператора энергии взаимодействия электронов:
здесь rij – расстояние

Используя (4.2), Хартри заменил оператор (4.1) на эффективный электронный потенциал, описывающий
Используя (4.2), Хартри заменил оператор (4.1) на эффективный электронный потенциал, описывающий

Для того, чтобы провести усреднение <…>, надо уже знать функции, описывающие
Для того, чтобы провести усреднение <…>, надо уже знать функции, описывающие

и строят оператор (h0i)ССП. Затем решают набор одно-электронных уравнений Хартри (4.3).
и строят оператор (h0i)ССП. Затем решают набор одно-электронных уравнений Хартри (4.3).

Многоэлектронная волновая функция ϕ(r) системы с замкнутой оболочкой, может быть описана
Многоэлектронная волновая функция ϕ(r) системы с замкнутой оболочкой, может быть описана

Метод Хартри-Фока
Fφi = εi φi
Как выглядят орбитали?
Аппроксимация многоэлектронной волновой функция единственным
Метод Хартри-Фока
Fφi = εi φi
Как выглядят орбитали?
Аппроксимация многоэлектронной волновой функция единственным

Приближение МО ЛКАО
Молекулярная Орбиталь –
Линейная Комбинация Атомных Орбиталей:
M атомных орбиталей
Приближение МО ЛКАО
Молекулярная Орбиталь –
Линейная Комбинация Атомных Орбиталей:
M атомных орбиталей

Выбор базисных АО
Должны давать хорошее приближение к истинной ВФ (например, возле
Выбор базисных АО
Должны давать хорошее приближение к истинной ВФ (например, возле

АО состоит из угловой и радиальной частей.
В качестве угловой части используются
АО состоит из угловой и радиальной частей.
В качестве угловой части используются

OCT (STO)
Функции STO – точные решения для радиальных частей орбиталей водородоподобного
OCT (STO)
Функции STO – точные решения для радиальных частей орбиталей водородоподобного

ОГТ (GTO)
Допускают быстрое вычисление интегралов (более удобны в вычислениях).
Не имеют физического
ОГТ (GTO)
Допускают быстрое вычисление интегралов (более удобны в вычислениях).
Не имеют физического

Общее резюме
Более физичные и «правильные» STO мало пригодны для вычислений.
Нефизичные с
Общее резюме
Более физичные и «правильные» STO мало пригодны для вычислений.
Нефизичные с

Метод Хартри-Фока-Рутана.
Базисные функции, обычно центрированы на атомах (Атомные Орбитали).
АО «образуют» базис
Метод Хартри-Фока-Рутана.
Базисные функции, обычно центрированы на атомах (Атомные Орбитали).
АО «образуют» базис

Уравнения Хартри-Фока. Подставим в них разложение МО по АО.
В обозначениях Дирака
умножим
Уравнения Хартри-Фока. Подставим в них разложение МО по АО.
В обозначениях Дирака умножим

Обозначим
Sνμ – матрица интегралов перекрывания, её элементы говорят о степени пространственного
Обозначим
Sνμ – матрица интегралов перекрывания, её элементы говорят о степени пространственного

Матричная форма уравнений Хартри-Фока-Рутана
F – матрица Фока с элементами Fνμ,
S –
Матричная форма уравнений Хартри-Фока-Рутана
F – матрица Фока с элементами Fνμ,
S –

При решении уравнений Хартри-Фока-Рутана, кроме интеграла перекрывания, необходимо вычислять большое число
При решении уравнений Хартри-Фока-Рутана, кроме интеграла перекрывания, необходимо вычислять большое число

Методы решения электронного уравнения Шредингера.
Неэмпирические (ab initio) методы расчета
(лабораторная работа №
Методы решения электронного уравнения Шредингера.
Неэмпирические (ab initio) методы расчета
(лабораторная работа №

Основная проблема – расчет интегралов на АО
Молекула СО – самый простой
Основная проблема – расчет интегралов на АО
Молекула СО – самый простой

Неэмпирические методы расчета:
Орбитали слейтеровского типа (STO) обычно
аппроксимируются орбиталями гауссова
Неэмпирические методы расчета:
Орбитали слейтеровского типа (STO) обычно
аппроксимируются орбиталями гауссова

Типы базисных наборов
(по числу базисных функций)
Минимальный базисный набор –
Типы базисных наборов
(по числу базисных функций)
Минимальный базисный набор –

Расширенный базисный набор
Используя две, три и т.д. функций на одну
Расширенный базисный набор Используя две, три и т.д. функций на одну

Точность ab initio методов
Ошибки ab initio расчета в основном обусловлены недостаточно
Точность ab initio методов
Ошибки ab initio расчета в основном обусловлены недостаточно

Полуэмпирические методы квантовой химии
Рассматриваем методы на основе приближения ХФ. В методе
Полуэмпирические методы квантовой химии
Рассматриваем методы на основе приближения ХФ. В методе

Общие требования к полуэмпирическим методам
Хотим…
Высокая скорость расчета БОЛЬШИХ систем.
Надежные результаты.
Легко интерпретируемые
Общие требования к полуэмпирическим методам
Хотим…
Высокая скорость расчета БОЛЬШИХ систем.
Надежные результаты.
Легко интерпретируемые

Общие приближения полуэмпирических методов
Валентное приближение.
Явно рассматриваются только валентные электроны
Минимальный
Общие приближения полуэмпирических методов
Валентное приближение.
Явно рассматриваются только валентные электроны
Минимальный
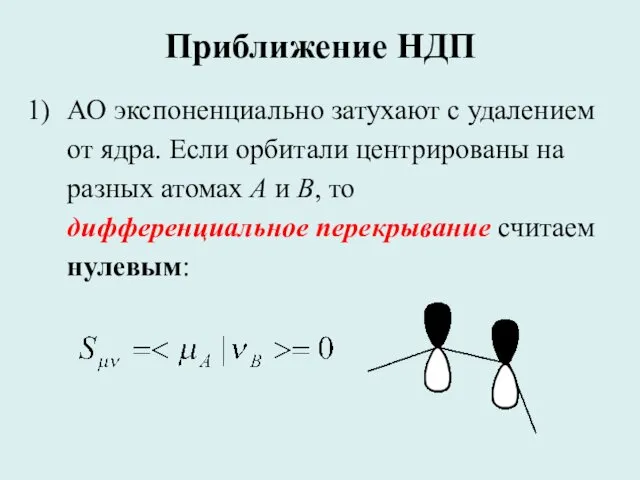
Приближение НДП
АО экспоненциально затухают с удалением от ядра. Если орбитали центрированы
Приближение НДП
АО экспоненциально затухают с удалением от ядра. Если орбитали центрированы
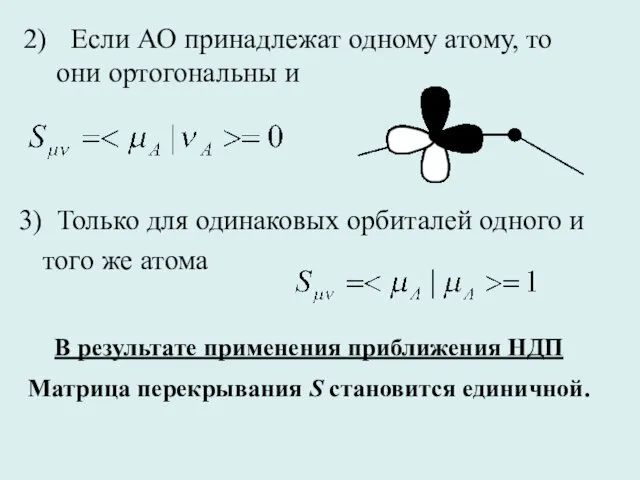
Если АО принадлежат одному атому, то они ортогональны и
3)
Если АО принадлежат одному атому, то они ортогональны и
3)
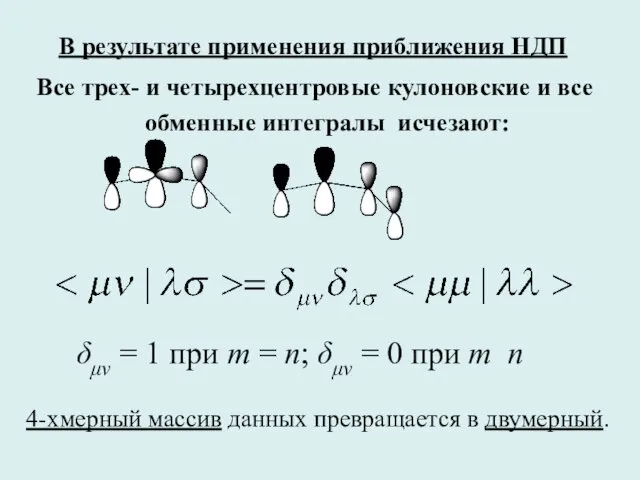
В результате применения приближения НДП
Все трех- и четырехцентровые кулоновские и все
В результате применения приближения НДП
Все трех- и четырехцентровые кулоновские и все

Плата за резкое упрощение.
Появляется большое количество параметров, численные значения которых
Плата за резкое упрощение.
Появляется большое количество параметров, численные значения которых

Метод MNDO (МПДП)
Modified Neglect of Diatomic Overlap (Модифицированное Пренебрежение Двухатомным Перекрыванием)
В
Метод MNDO (МПДП)
Modified Neglect of Diatomic Overlap (Модифицированное Пренебрежение Двухатомным Перекрыванием)
В

Методы AM1 и PM3
Специально параметризованы для описания водородных связей.
АМ1 (Austin Model
Методы AM1 и PM3
Специально параметризованы для описания водородных связей.
АМ1 (Austin Model

Полуэмпирические методы, учитывающие электронную корреляцию, CNDO/S и INDO/S
Они параметризованы для расчетов
Полуэмпирические методы, учитывающие электронную корреляцию, CNDO/S и INDO/S
Они параметризованы для расчетов
 Двоичные сумматоры
Двоичные сумматоры Окружной конкурс исследовательских работ. Номинация: физико–математическая. Укротитель волшебного пламени
Окружной конкурс исследовательских работ. Номинация: физико–математическая. Укротитель волшебного пламени Контрольное задание №1. Определение типов темперамента
Контрольное задание №1. Определение типов темперамента Методы управления персоналом
Методы управления персоналом СТРАТЕГИЯ МЕТОДА ТРЕХ МЫСЛИТЕЛЬНЫХ СТУЛЬЕВ
СТРАТЕГИЯ МЕТОДА ТРЕХ МЫСЛИТЕЛЬНЫХ СТУЛЬЕВ Роль семьи в нравственном воспитании подрастающего поколения, в ориентировании его на ценности гражданского общества
Роль семьи в нравственном воспитании подрастающего поколения, в ориентировании его на ценности гражданского общества Теплогидравлика новой конструкции. ТВС для реакторов IV поколения со сверхкритическими параметрами теплоносителя
Теплогидравлика новой конструкции. ТВС для реакторов IV поколения со сверхкритическими параметрами теплоносителя Дискинезии желчевыводящих путей у детей
Дискинезии желчевыводящих путей у детей Презентация для урока по теме Тепло в атмосфере 6 класс
Презентация для урока по теме Тепло в атмосфере 6 класс Металлокерамикалық көпіртәрізді протездің клиникалық реставрациясы
Металлокерамикалық көпіртәрізді протездің клиникалық реставрациясы Самоподготовка
Самоподготовка Неопределенный интеграл. Методы интегрирования
Неопределенный интеграл. Методы интегрирования Особенности учета дров. Правила укладки и обмера, единицы учета, полнодревесность поленниц
Особенности учета дров. Правила укладки и обмера, единицы учета, полнодревесность поленниц Загадки и их виды
Загадки и их виды Генеалогические карточки
Генеалогические карточки Европейский иррационализм
Европейский иррационализм Маркетинг ресторанных услуг
Маркетинг ресторанных услуг Роль машиностроения в развитии экономики России
Роль машиностроения в развитии экономики России Волейбол. Встречная передача и прием мяча сверху и снизу. Верхняя прямая подача, прием с подачи
Волейбол. Встречная передача и прием мяча сверху и снизу. Верхняя прямая подача, прием с подачи Сварочное производство (тема 1.3)
Сварочное производство (тема 1.3) Классный час. Тема: Природа и здоровье человека
Классный час. Тема: Природа и здоровье человека Викторина. Своя игра.
Викторина. Своя игра. Требования к руководству испытательных и калибровочных лабораторий (ISO/IEC 17025)
Требования к руководству испытательных и калибровочных лабораторий (ISO/IEC 17025) Судебная система в Российской Федерации
Судебная система в Российской Федерации Технология приготовления хлебобулочного изделия хлеба Тольяттинский и мучного кондитерского изделия Рожки с повидлом
Технология приготовления хлебобулочного изделия хлеба Тольяттинский и мучного кондитерского изделия Рожки с повидлом Программа развития для ДОУ
Программа развития для ДОУ Проектирование и разработка автоматизированной информационной системы для гостиничного предприятия
Проектирование и разработка автоматизированной информационной системы для гостиничного предприятия Устройство и принцип действия транзистора
Устройство и принцип действия транзистора