- Химическая термодинамика

Содержание
- 2. Термодинамика – это наука, изучающая переходы энергии из одной формы в др., от одних частей системы
- 3. Хим. в-ва, входящие в состав системы, явл-ся её составными частями или компонентами. Системы м.б. одно-, двух-
- 4. Гомогенные системы – это смеси газов, истинные растворы (жидкие или твердые) и др. Гетерогенные системы состоят
- 5. В ходе различных превращений система переходит из одного энергетического состояния в другое. То или иное состояние
- 6. Др. параметры, зависящие от основных, наз-ся ТД функциями состояния системы. В химии наиболее часто используются :
- 7. Внутренняя энергия системы (U) – это полная энергия системы, включающая кине-тическую энергию всех видов движения молекул,
- 8. Запас внутр. энергии системы зависит от параметров состояния системы, природы в-ва и прямо пропорционален массе вещества.
- 9. Изм-ние внутр. энергии системы (ΔU), как и изм-ние любой ТД функции, опр-ся разностью её величин в
- 10. При переходе неизолированной системы из одного состояния в другое изменение её внутренней энергии осуществляется путём обмена
- 11. Рис.1. Изменение внутренней энергии При р = const теплота Qp идёт на увеличение запаса внутренней энергии
- 12. Ур-ние: Qр = ΔU + А выражает суть первого закона ТД: сумма изменений внутренней энергии и
- 13. Это ещё одна важная т.д. ф-ция состояния системы: энтальпия или теплосодержание. Тогда Qp = ΔU +
- 14. В изохорических условиях (V = const и ΔV = 0) вся подведённая к системе теплота (Qv)
- 15. Теплоты хим р-ций, протекающих в изохорно-изотермических и изобарно-изотермических усло-виях, называют тепловыми эффектами. (Дж/моль или кДж/моль). Тепловые
- 16. ТЕРМОХИМИЯ. ТЕРМОХИМ. УРАВНЕНИЯ. ТЕРМОХИМИЧЕСКИЕ РАСЧЁТЫ Раздел химии и хим. ТД, занятый расчётами тепловых эффектов, наз-ся термохимией.
- 17. Поскольку абсолютные значения энергии (т.д. ф-ций) измерить принципиально невозможно, то для проведения термохим. расчётов вводят специальные
- 18. При составлении термохим. ур-ний обр-ния 1 моль некоторых веществ возможно применение нецелочисленных коэффициентов. Поскольку условия получения
- 19. Стандартные условия стандартное давление – 0,1 МПа или 1 атм стандартная температура – 25°C или 298
- 20. Т.к. тепловой эффект р-ций зависит от агре-гатного состояния в-в, то в термохим. ур-ниях указывается и их
- 21. В основе термохимических расчётов реакций лежит закон Гесса (1836 – 1841): Тепловой эффект реакции (ΔНр) не
- 22. 1. Тепловой эффект реакции равен сумме теплот обр-ния продуктов р-ции (Σν2ΔН0обр.прод.) за вычетом суммы теплот обр-ния
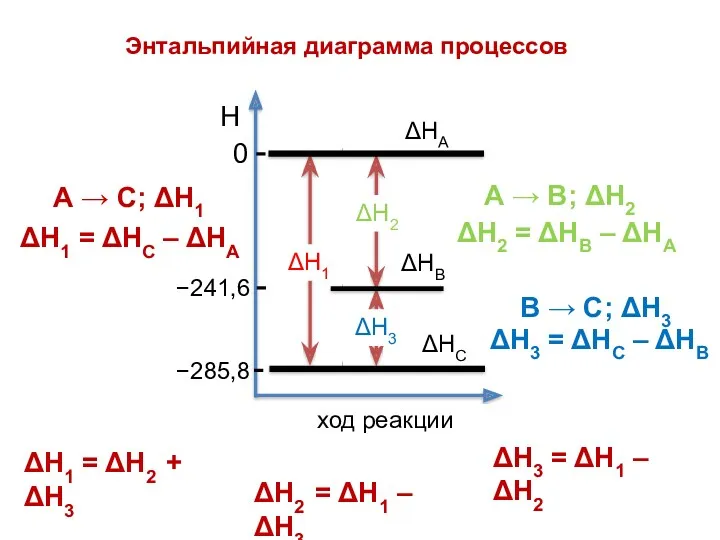
- 23. ΔН2 ΔН1 Н ΔН3 ход реакции 0 −285,8 −241,6 ΔН3 = ΔН1 – ΔН2 ΔН1 =
- 24. Н2 (г) + ½О2(г) = Н2О(ж); ΔН1 ΔН1 = ΔН0обрН2О(ж) – (ΔН0обрН2 (г) + ½ΔН0обрО2 (г))
- 25. Теплота обр-ния жидкой воды не зависит от способа её получения: 1) сжигая Н2 и О2 (ΔΗ1)
- 26. Пример вычитания термохимических уравнений : _ Н2(г) + О2(г) = Н2О(ж); ΔН1 Н2(г) + О2(г) =
- 27. ЭНТРОПИЯ По з-ну сохранения энергии система может самопроизвольно совершать работу только за счёт собственной энергии, т.е.
- 28. Имеется несколько его формулировок. 1 – Постулат Клаузиуса: теплота не переходит от холодного тела к горячему.
- 29. Из постулатов следует, что в обратимом процессе : Это эквивалентно утверждению, что dQ/T есть дифференциал нек-рой
- 30. Л.Больцман (1877): Т.д. вероятность W состояния системы – это число микросостояний, реализующих данное макросостояние: S =
- 31. S1 S2 ΔН = 0 S1 S2 Рассмотрим изолированную систему из двух газов . ΔS =
- 32. Действующая сила процесса связана со стремлением ТД систем к самопроизвольному ув-нию степени хаотичности или ув-нию энтропии.
- 33. С понижением т-ры энтропия в-ва ум-ся (ум-ся скорость движения частиц, число микро-состояний и Т.Д. вероятность W
- 34. ΔS хим. реакции также не зависит от пути процесса, а определяется лишь энтропией начального и конечного
- 35. Энтропийный фактор является одной из двух движущих сил процессов и должен иметь размер энергии. Для этого
- 36. ЭНЕРГИЯ ГИББСА С учетом одновременного действия двух противоположных факторов движущей силой для р-ций, протекающих при P,T=const,
- 37. ΔG ΔG > 0 ΔG = 0 реакция термодинамически возможна При постоянной т-ре и давлении хим.
- 38. Энергия Гиббса связана с энтальпией, энтропией и температурой: G = H – Т⋅S. Её изм-ние ΔG:
- 39. Ι ΙΙΙ ΙΙ ΙV I. если ΔН 0, то ΔG II. Если ΔН > 0, а
- 40. Стандартная энергия Гиббса обр-ния в-ва (ΔG0обр. 298) – изм-ние энергии Гиббса в р-ции обр-ния 1 моль
- 42. Скачать презентацию

Термодинамика – это наука, изучающая переходы энергии из одной формы в
Термодинамика – это наука, изучающая переходы энергии из одной формы в

Хим. в-ва, входящие в состав системы, явл-ся её составными частями или
Хим. в-ва, входящие в состав системы, явл-ся её составными частями или

Гомогенные системы – это смеси газов, истинные растворы (жидкие или твердые)
Гомогенные системы – это смеси газов, истинные растворы (жидкие или твердые)

В ходе различных превращений система переходит из одного энергетического состояния в
В ходе различных превращений система переходит из одного энергетического состояния в

Др. параметры, зависящие от основных, наз-ся ТД функциями состояния системы.
В
Др. параметры, зависящие от основных, наз-ся ТД функциями состояния системы.
В

Внутренняя энергия системы (U) – это полная энергия системы, включающая кине-тическую
Внутренняя энергия системы (U) – это полная энергия системы, включающая кине-тическую

Запас внутр. энергии системы зависит от параметров состояния системы, природы в-ва
Запас внутр. энергии системы зависит от параметров состояния системы, природы в-ва

Изм-ние внутр. энергии системы (ΔU), как и изм-ние любой ТД функции,
Изм-ние внутр. энергии системы (ΔU), как и изм-ние любой ТД функции,

При переходе неизолированной системы из одного состояния в другое изменение её
При переходе неизолированной системы из одного состояния в другое изменение её
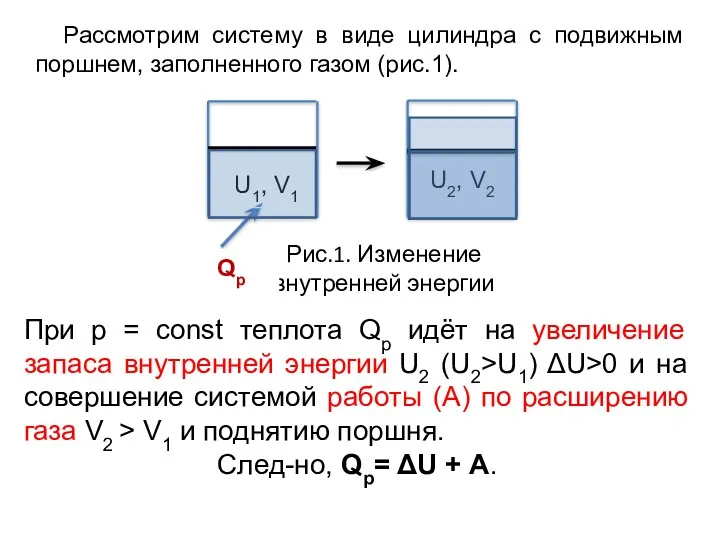
Рис.1. Изменение
внутренней энергии
При р = const теплота Qp идёт на
Рис.1. Изменение
внутренней энергии
При р = const теплота Qp идёт на

Ур-ние: Qр = ΔU + А выражает суть первого закона ТД:
Ур-ние: Qр = ΔU + А выражает суть первого закона ТД:

Это ещё одна важная т.д. ф-ция состояния системы: энтальпия или теплосодержание.
Это ещё одна важная т.д. ф-ция состояния системы: энтальпия или теплосодержание.

В изохорических условиях (V = const и ΔV = 0) вся
В изохорических условиях (V = const и ΔV = 0) вся

Теплоты хим р-ций, протекающих в изохорно-изотермических и изобарно-изотермических усло-виях, называют тепловыми
Теплоты хим р-ций, протекающих в изохорно-изотермических и изобарно-изотермических усло-виях, называют тепловыми

ТЕРМОХИМИЯ. ТЕРМОХИМ. УРАВНЕНИЯ.
ТЕРМОХИМИЧЕСКИЕ РАСЧЁТЫ
Раздел химии и хим. ТД, занятый расчётами
ТЕРМОХИМИЯ. ТЕРМОХИМ. УРАВНЕНИЯ.
ТЕРМОХИМИЧЕСКИЕ РАСЧЁТЫ
Раздел химии и хим. ТД, занятый расчётами

Поскольку абсолютные значения энергии (т.д. ф-ций) измерить принципиально невозможно, то для
Поскольку абсолютные значения энергии (т.д. ф-ций) измерить принципиально невозможно, то для

При составлении термохим. ур-ний обр-ния
1 моль некоторых веществ возможно применение
При составлении термохим. ур-ний обр-ния 1 моль некоторых веществ возможно применение

Стандартные условия
стандартное давление – 0,1 МПа или 1 атм
стандартная
Стандартные условия
стандартное давление – 0,1 МПа или 1 атм
стандартная

Т.к. тепловой эффект р-ций зависит от агре-гатного состояния в-в, то в
Т.к. тепловой эффект р-ций зависит от агре-гатного состояния в-в, то в

В основе термохимических расчётов реакций лежит закон Гесса (1836 – 1841):
Тепловой
В основе термохимических расчётов реакций лежит закон Гесса (1836 – 1841):
Тепловой

1. Тепловой эффект реакции равен сумме теплот обр-ния продуктов р-ции (Σν2ΔН0обр.прод.)
1. Тепловой эффект реакции равен сумме теплот обр-ния продуктов р-ции (Σν2ΔН0обр.прод.)
ΔН2
ΔН1
Н
ΔН3
ход реакции
0
−285,8
−241,6
ΔН3 = ΔН1 – ΔН2
ΔН1 =
ΔН2
ΔН1
Н
ΔН3
ход реакции
0
−285,8
−241,6
ΔН3 = ΔН1 – ΔН2
ΔН1 =
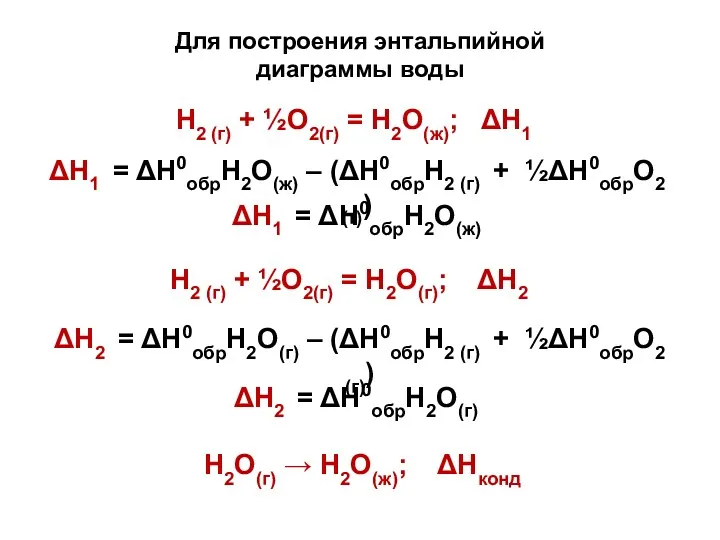
Н2 (г) + ½О2(г) = Н2О(ж); ΔН1
ΔН1 = ΔН0обрН2О(ж) – (ΔН0обрН2
Н2 (г) + ½О2(г) = Н2О(ж); ΔН1
ΔН1 = ΔН0обрН2О(ж) – (ΔН0обрН2
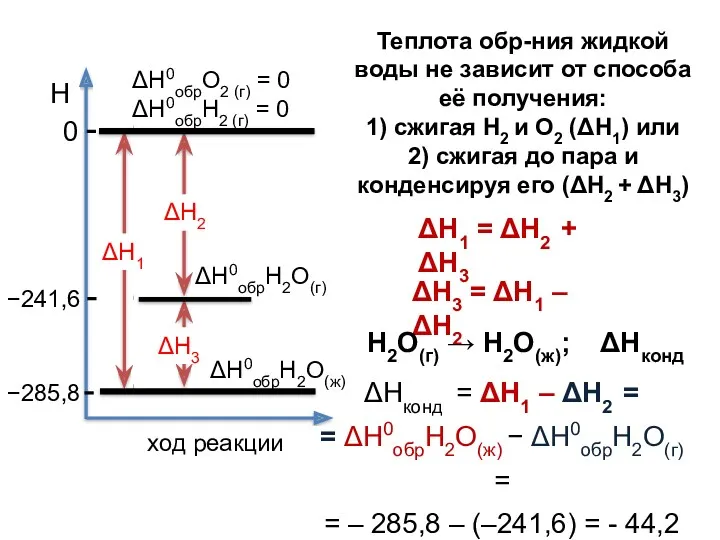
Теплота обр-ния жидкой воды не зависит от способа её получения:
1)
Теплота обр-ния жидкой воды не зависит от способа её получения:
1)
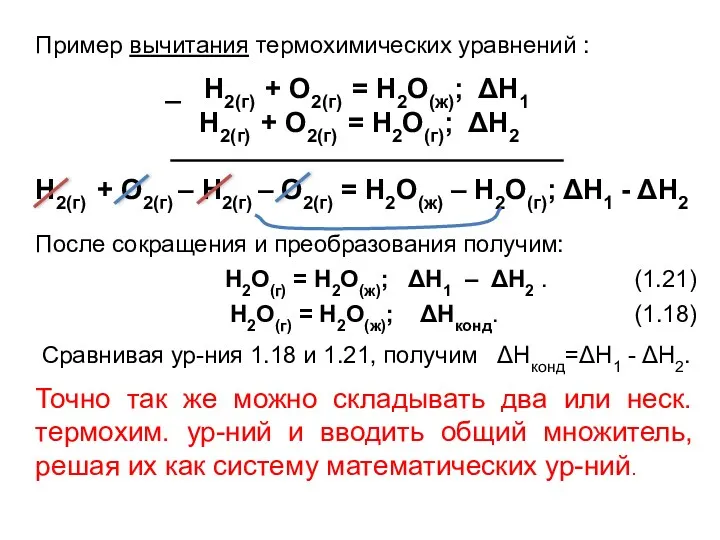
Пример вычитания термохимических уравнений :
_ Н2(г) + О2(г) = Н2О(ж);
Пример вычитания термохимических уравнений :
_ Н2(г) + О2(г) = Н2О(ж);

ЭНТРОПИЯ
По з-ну сохранения энергии система может самопроизвольно совершать работу только за
ЭНТРОПИЯ
По з-ну сохранения энергии система может самопроизвольно совершать работу только за
Имеется несколько его формулировок.
1 – Постулат Клаузиуса: теплота не переходит
Имеется несколько его формулировок.
1 – Постулат Клаузиуса: теплота не переходит

Из постулатов следует, что в обратимом процессе :
Это эквивалентно утверждению, что
Из постулатов следует, что в обратимом процессе :
Это эквивалентно утверждению, что

Л.Больцман (1877): Т.д. вероятность W состояния системы – это число микросостояний,
Л.Больцман (1877): Т.д. вероятность W состояния системы – это число микросостояний,
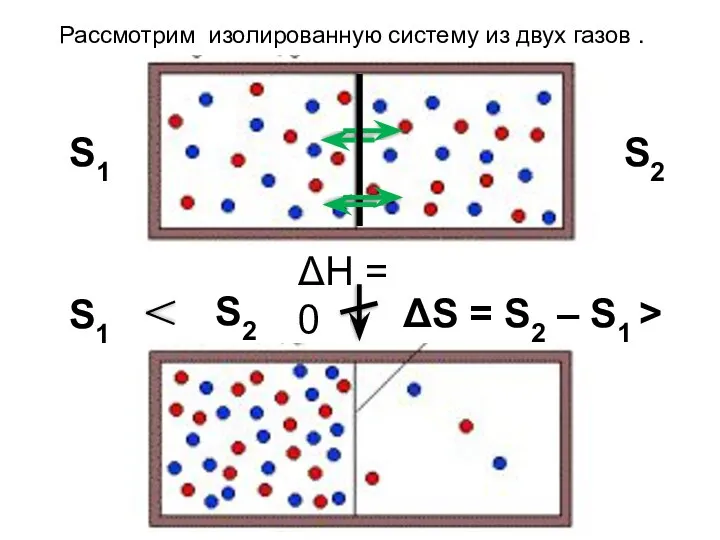
S1
S2
ΔН = 0
S1
S2
Рассмотрим изолированную систему из двух газов .
ΔS = S2
S1
S2
ΔН = 0
S1
S2
Рассмотрим изолированную систему из двух газов .
ΔS = S2

Действующая сила процесса связана со стремлением ТД систем к самопроизвольному ув-нию
Действующая сила процесса связана со стремлением ТД систем к самопроизвольному ув-нию
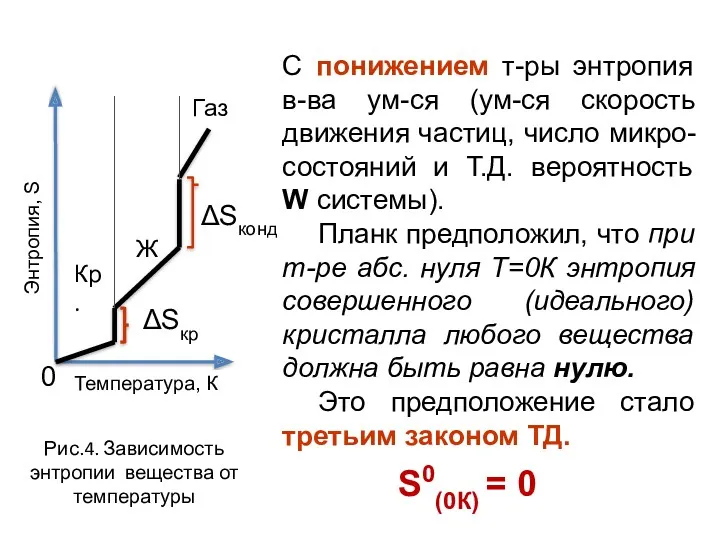
С понижением т-ры энтропия в-ва ум-ся (ум-ся скорость движения частиц, число
С понижением т-ры энтропия в-ва ум-ся (ум-ся скорость движения частиц, число

ΔS хим. реакции также не зависит от пути процесса, а определяется
ΔS хим. реакции также не зависит от пути процесса, а определяется

Энтропийный фактор является одной из двух движущих сил процессов и должен
Энтропийный фактор является одной из двух движущих сил процессов и должен

ЭНЕРГИЯ ГИББСА
С учетом одновременного действия двух противоположных факторов движущей силой для
ЭНЕРГИЯ ГИББСА
С учетом одновременного действия двух противоположных факторов движущей силой для

ΔG< 0
ΔG > 0
ΔG = 0
реакция термодинамически возможна
При постоянной т-ре и
ΔG< 0
ΔG > 0
ΔG = 0
реакция термодинамически возможна
При постоянной т-ре и

Энергия Гиббса связана с энтальпией, энтропией и температурой: G = H
Энергия Гиббса связана с энтальпией, энтропией и температурой: G = H
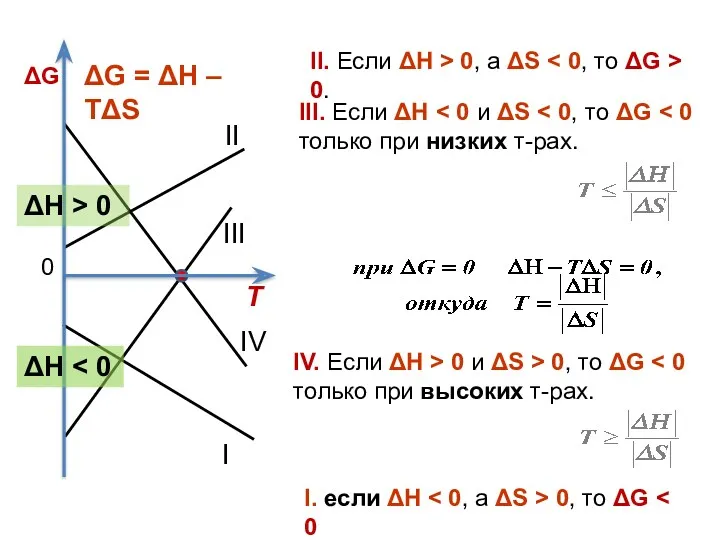
Ι
ΙΙΙ
ΙΙ
ΙV
I. если ΔН < 0, а ΔS > 0, то ΔG
Ι
ΙΙΙ
ΙΙ
ΙV
I. если ΔН < 0, а ΔS > 0, то ΔG

Стандартная энергия Гиббса обр-ния в-ва (ΔG0обр. 298) – изм-ние энергии Гиббса
Стандартная энергия Гиббса обр-ния в-ва (ΔG0обр. 298) – изм-ние энергии Гиббса
 Карбоновые кислоты – союз двух групп (урок-исследование химических свойств карбоновых кислот)
Карбоновые кислоты – союз двух групп (урок-исследование химических свойств карбоновых кислот) Аллотропные модификации алмаза
Аллотропные модификации алмаза Вещества и тела. Состояния веществ. Смеси
Вещества и тела. Состояния веществ. Смеси Хром. Свойства
Хром. Свойства Кремний и его соединения
Кремний и его соединения D-элементы: хром, молибден, вольфрам
D-элементы: хром, молибден, вольфрам Реакционная способность твердых тел и способы ее регулирования
Реакционная способность твердых тел и способы ее регулирования Гибкость цепи полимеров
Гибкость цепи полимеров Общая характеристика неметаллов
Общая характеристика неметаллов Реакция Бэйлиса Хиллмана-Мориты
Реакция Бэйлиса Хиллмана-Мориты Аминокислоты. Пептиды. Белки
Аминокислоты. Пептиды. Белки Коррозия металлов
Коррозия металлов Коррозия металлов
Коррозия металлов Preparation for COP
Preparation for COP Экспериментальные методы измерения изотерм адсорбции. Лекция 4
Экспериментальные методы измерения изотерм адсорбции. Лекция 4 Закон сохранения массы веществ. Химические уравнения
Закон сохранения массы веществ. Химические уравнения Титан және оның қорытпалары. Титаннан жасалған құралдар
Титан және оның қорытпалары. Титаннан жасалған құралдар Озон
Озон Соединения азота. Оксиды азота
Соединения азота. Оксиды азота Водород. Общая характеристика по плану
Водород. Общая характеристика по плану Исследовательский проект определение качества питьевой воды в домашних условиях
Исследовательский проект определение качества питьевой воды в домашних условиях Классификация конструкционных материалов. Особенности атомно-кристаллического строения металлов. Лекции №1
Классификация конструкционных материалов. Особенности атомно-кристаллического строения металлов. Лекции №1 Разделение углеводородных газов
Разделение углеводородных газов Химические реакции
Химические реакции Каменный уголь. Переработка и применение угля
Каменный уголь. Переработка и применение угля Гидролиз неорганических соединений
Гидролиз неорганических соединений Водород. Химический элемент
Водород. Химический элемент Металдар мен бейметалдардың салыстырмалы сипаттамасы
Металдар мен бейметалдардың салыстырмалы сипаттамасы