Теоретические основы химической технологии переработки природных энергоносителей и углеводородных материалов презентация
- Теоретические основы химической технологии переработки природных энергоносителей и углеводородных материалов

Содержание
- 2. Лекция 1. Введение Основные цели и задачи курса В данном курсе рассматриваются основные понятия фундаментальных наук
- 3. Некоторые положения и выводы изучаемого курса не будут носить общего теоретического характера, так как речь пойдет
- 4. Конечный результат химической реакции процесса может быть описан обычными химическими уравнениями, не раскрывающими пути протекания реакции,
- 5. Механизм процесса химической реакции характеризуется способами разрыва и образования связей Существуют два основных способа разрыва или
- 6. Скорости прямой (или обратной) реакций в общем виде описываются законом действующих масс: где w – скорость
- 7. Для разрыва связи с образованием свободных радикалов требуется меньше энергии, чем для разрыва по карбоний-ионному механизму,
- 8. Лекция 2. Образование и свойства радикалов Радикалом называется активная промежуточная частица, имеющая во внешней оболочке неспаренные
- 9. Энергия разрыва их соответственно 360 и 410 кДж/моль. Следовательно . При 600 0С . Т.е. распад
- 10. В молекулах алкенов (олефинов) одинарные связи у атома углерода с двойной связью (α-связи) значительно прочнее, чем
- 11. Если двойная связь сопряжённая (т.е. связь находящаяся в ß -положении, по сравнению с двойной связью, тоже
- 12. Прочность связи С-Н в циклоалкановых (нафтеновых) кольцах такая же, как связь Свтор-Н в алканах. Связь С-С
- 13. Свойства радикалов (продолжение) 4) Энергия связи С-Н у первого атома углерода в цепи выше, чем у
- 14. Лекция 3. Реакции радикалов. Замещение и присоединение В силу своей природы радикалы вступают в реакции с
- 15. При наличии в молекуле связей С-Н с различной прочностью, реакция радикала с молекулой происходит селективно (избирательно).
- 16. Реакции типа вследствие энергетических (высокой энергии активации) и стерических (малый стерический множитель, учитывающийся в предэкспоненциальном множителе,
- 17. При реакции затрачивается 249 кДж/моль на разрыв π-связи и выделяется в зависимости от вида R и
- 18. Лекция 4. Реакции радикалов. Распад, изомеризация, рекомбинация и диспропорционирование, цепные реакции свободных радикалов Распад Радикалы могут
- 19. Когда радикал может распасться по нескольким направлениям, то вероятность распада по различным связям определяется в первую
- 20. Бирадикалы распадаются так же, как и обычные радикалы, но свободные валентности при этом исчезают: Изомеризация При
- 21. Алкановые радикалы не способны к 1,2-изомеризации. Однако для аренов наблюдается 1,2-перход, связанный с внутримолекулярным присоединением. Рекомбинация
- 22. Цепные реакции свободных радикалов Образовавшийся радикал при распаде или реакции с молекулой превращается в другой радикал;
- 23. Суммарная реакция чередующихся элементарных реакций стадии звено цепи даёт уравнение распада этана: Некоторое конечное число звеньев
- 24. Лекция 5. Пиролиз в газовой фазе. Термолиз алканов и алкенов Пиролизом называется процесс высокотемпературного (750-8000С) термолиза
- 25. Термолиз алканов Термолиз алканов приводит к образованию более термостабильных алканов и алкенов. Наиболее термостабильным из алканов
- 26. Помимо образующихся в больших количествах этилена и водорода (из 1 моль этана теоретически получается 1 моль
- 27. В процессе пиролиза н-бутана преобладают две реакции его распада: 1) ; 2) . Нормальные парафиновые углеводороды
- 28. Самым термостабильным из гомологического ряда алкенов является этилен (деструкция начинается при 6600С). По термической стабильности он
- 29. Пропилен по термической стабильности уступает этилену и при термолизе образует метан и этилен: Термолиз бутиленов приводит
- 30. Лекция 6. Пиролиз в газовой фазе. Термолиз нафтенов и ароматики Термолиз нафтенов Нафтены (циклопарафины) при термолизе
- 31. Дегидрирование незамещённых циклоалканов по цепному механизму не происходит, т.к. распад с образование бирадикалов протекает со скорость
- 32. Механизм превращения циклогексана при термолизе:
- 33. Механизм превращения циклопентана при термолизе: Пироуглерод откладывается в трубах печи, ухудшая их теплопроводность. Поэтому нафтеновые углеводороды
- 34. Термолиз ароматических углеводородов Термоустойчивасть аренов зависит от наличия алкильных цепей в их молекулах. Связи, сопряжённые с
- 35. В результате конденсации нафталина образуется более высококонденсированная ароматика: нафталин динафтил перилен Ароматические углеводороды накапливаются в жидких
- 36. 4) Алкены полимеризуются и вступают в реакции деструктивной полимеризации, также возможны реакции циклизации; 5) Цикланы и
- 37. Лекция 7. Жидкофазный термолиз Жидкофазный термолиз имеет место в таких термодеструктивных процессах нефтепереработки, как термический крекинг,
- 38. «Клеточный эффект» Сущность «клеточного эффекта» заключается в том, что при распаде молекулы с образованием радикала в
- 39. Анализ термического превращения углеводородов на примере процесса замедленного коксования Одним из вариантов переработки ТНО является процесс
- 40. На каждой стадии образуются помимо продуктов уплотнения газы и более низкомолекулярные жидкости, чем образующиеся промежуточные продукты
- 41. На скорость термодеструктивных превращений ТНО существенно влияет растворяющая способность дисперсионной среды (ДС), которая определяет значение так
- 42. Принципиальная схема процесса замедленного коксования
- 43. К основным закономерностям процесса коксования относятся 1. Сырье коксования должно быть с большим содержанием смол и
- 44. 5. С повышением давления часть испарившихся углеводородов переходит в жидкую ДС, пороговая концентрация асфальтенов при этом
- 45. Лекция 8. Образование и свойства карбоний-ионов Как установлено выше, образование ионов при разрыве ковалентной связи (гетеролитическом
- 46. По природе промежуточного химического взаимодействия реагирующих химических веществ: 1) Гомолитический – когда взаимодействие идёт по гомолитическому
- 47. Энергия, необходимая для образования К-И, возрастает с увеличением числа атомов водорода, связанных с атомом углерода, от
- 48. При этом скорее образуется вторичный К-И, нежели первичный, т.к. он более устойчив: В результате взаимодействия олефина
- 49. 2) Неклассический способ Кислоты бывают настолько сильными, что они способны протонировать даже протонированный углеводород, благодаря этому
- 50. Таким образом, образование К-И по классическому способу происходит в соответствии с протонной теорией Бренстеда, где кислоты
- 51. Лекция 9. Реакции карбоний-ионов. Замещение и изомеризация Замещение (перенос гидрид-иона) Перенос гидрид-иона наблюдается при взаимодействии К-И
- 52. Бензин КК будет обладать большим октановым числом, так как содержание изопарафиновых и ароматических углеводородов больше, чем
- 53. Перегруппировка (изомеризация) Важной реакцией К-И является реакция перегруппировки путём сдвига атома водорода или атома углерода. Изомеризация
- 54. Изомеризация насыщенных углеводородов протекает через образование таких же К-И, но на первой стадии необходим отрыв гидрид-иона.
- 55. Лекция 10. Реакции карбоний-ионов. Присоединение, распад и конденсация Присоединение К-И, как и радикалы, вступают в реакцию
- 56. Алкилирование изопарафинов олефинами требует использования более сильных кислот, например концентрированной H2SO4 или HF. Реакция включает перенос
- 57. Распад (крекинг) К-И, как и радикалы, с большой скоростью подвергаются реакции распада (обратной реакции полимеризации), которая
- 58. Продолжение крекинга прямой цепи по ß-связям приводит к образованию пропилена с высоким выходом. В случае этого
- 60. После того как ароматические углеводороды образовались, они могут вступать в реакцию конденсации с образованием углеводородов более
- 61. Лекция 11. Характеристика катализа и катализаторов Большинство промышленных процессов основано на каталитических реакциях, и их совершенствование
- 62. Необходимое условие катализа Чтобы каталитическая реакция осуществлялась, энергия активации её должна быть ниже, чем некаталитической: На
- 63. Активность и селективность катализатора Скорость данной реакции в присутствии различных катализаторов характеризуется их активностью относительно данной
- 64. Стабильность катализатора Стабильностью катализатора называется его способность сохранять активность во времени. Жидкий катализатор дезактивируется из-за накопления
- 65. Химические изменения катализатора в процессе его работы вызываются хемосорбцией на его поверхности примесей к сырью. При
- 66. Кислотно-основный катализ В кислотно-основных реакциях промежуточные активные частицы – ионы, и катализатор инициирует их образование в
- 67. Окислительно-восстановительный катализ В окислительно-восстановительных реакциях промежуточные частицы – радикалоподобные нейтральные образования, связанные с активными центрами катализатора
- 68. Координационно-комплексный (бифункциональный) катализ В координационно-комплексном катализе при образовании комплекса реагента с катализатором происходит поляризация или ионизация
- 69. Лекция 12. Алюмосиликатные катализаторы Промышленное значение имеют катализаторы трёх типов: Природные активированные алюмосиликаты; Синтетические аморфные алюмосиликаты;
- 70. Катализаторы этого типа обладают слабой устойчивостью к действию высоких температур. Железо, содержащееся в их структуре, катализирует
- 71. Протонные кислотные центры образуются, например, при замещении алюминием атомов кремния в структуре силикагеля. При этом атом
- 72. Апротонные кислотные центры могут иметь следующую структуру: Атом алюминия в такой структуре является акцептором электронной пары,
- 73. Химический состав алюмосиликатов описывается соотношением SiO2 : Al2O3, называемым силикатным модулем. С изменением соотношения SiO2 :
- 74. Синтетические кристаллические алюмосиликаты Цеолиты (от греч. цео - кипящий, литос - камень) – кристаллические алюмосиликаты, общая
- 75. Цеолиты типа X и Y имеют кристаллическую структуру, образованную тетраэдрами SiO4 и AlO4: Причём, каждый атом
- 76. Атомы алюминия несут одиночный отрицательный заряд, вследствие чего алюмосиликатная решётка заряжена отрицательно. Отрицательные заряды решётки компенсируются
- 77. В результате объединения множества суперклеток (в фожазите их восемь) в регулярную систему формируется элементарная ячейка цеолита:
- 78. Промышленные катализаторы Катализаторы современных крупнотоннажных процессов каталитического крекинга, осуществляемых при высоких температурах (500-800оС) в режиме интенсивного
- 79. 2) Активного компонента – цеолита; На поверхности цеолитсодержащих катализаторов наблюдаются два типа кислотных центров: Бренстедовский (БКЦ)
- 80. Перечень наиболее типичных вспомогательных добавок: В качестве промотора, идентифицирующего регенерацию закоксованного катализатора, чаще всего применяют платину,
- 81. Лекция 13. Процессы, идущие по бифункциональному катализу: каталитический риформинг и изомеризация Радикальные реакции могут катализироваться некоторыми
- 82. Каталитический риформинг Целью процесса является повышение октанового числа низкооктановых бензиновых фракций. Целевая реакция – ароматизация углеводородов.
- 83. Дегидроизомеризация 5-членных цикланов фактически разбивается на два процесса, а именно: изомеризацию, протекающую на кислотных центрах и
- 84. Деалкилирование алкиларенов протекает на кислотных центрах: Коксообразование также протекает на кислотных центрах: В условиях каталитического риформинга
- 85. Изомеризация в бифункциональном катализе На бифункциональных катализаторах, обладающих дегидро-гидрирующей и кислотной активностью, изомеризация протекает по следующей
- 86. Давление не оказывает существенного влияния на стадию дегидрирования и гидрирования. Повышение давления водорода приводит к затруднению
- 87. Лекция 14. Процессы, идущие по бифункциональному катализу: гидрокрекинг Гидрокрекинг – каталитический процесс переработки нефтяных дистиллятов (реже
- 88. Гидрокрекинг алканов на катализаторах с высокой кислотной активностью протекает по карбкатионному механизму: Циклоалканы с длинными алкильными
- 89. Механизм деалкилирования может быть описан следующей схемой:
- 90. И далее возможны два пути протекания реакции. Первый с образованием в конечном итоге нормального парафина: Второй
- 91. Лекция 15. Процессы, идущие по бифункциональному катализу: гидроочистка Гидроочистка – процесс удаления из нефтепродуктов гетероатомов в
- 92. Гидроочистка серусодержащих соединений Серусодержащие соединения, имеющиеся в нефтепродукте подвергаются следующим изменениям: Меркаптаны гидрируются до сервоводорода и
- 93. Тетрагидротиофены гидрируются с образованием соответствующих алифатических углеводородов: Тиофены дают такие же продукты, как и тетрагидротиофены: Скорость
- 94. На кинетику реакций гидрогенолиза сильное влияние оказывает тип и строение гетероорганических соединений. Скорость гидрогенолиза серусодержащих соединений
- 96. Скачать презентацию

Лекция 1. Введение
Основные цели и задачи курса
В данном курсе рассматриваются
Лекция 1. Введение
Основные цели и задачи курса
В данном курсе рассматриваются

Некоторые положения и выводы изучаемого курса не будут носить общего теоретического
Некоторые положения и выводы изучаемого курса не будут носить общего теоретического

Конечный результат химической реакции процесса может быть описан обычными химическими уравнениями,
Конечный результат химической реакции процесса может быть описан обычными химическими уравнениями,

Механизм процесса химической реакции характеризуется
способами разрыва и образования связей
Существуют два основных
Механизм процесса химической реакции характеризуется
способами разрыва и образования связей
Существуют два основных

Скорости прямой (или обратной) реакций в общем виде описываются законом действующих
Скорости прямой (или обратной) реакций в общем виде описываются законом действующих

Для разрыва связи с образованием свободных радикалов требуется меньше энергии, чем
Для разрыва связи с образованием свободных радикалов требуется меньше энергии, чем

Лекция 2.
Образование и свойства радикалов
Радикалом называется активная промежуточная частица,
Лекция 2.
Образование и свойства радикалов
Радикалом называется активная промежуточная частица,

Энергия разрыва их соответственно 360 и 410 кДж/моль. Следовательно
.
При
Энергия разрыва их соответственно 360 и 410 кДж/моль. Следовательно
.
При

В молекулах алкенов (олефинов) одинарные связи у атома углерода с двойной
В молекулах алкенов (олефинов) одинарные связи у атома углерода с двойной
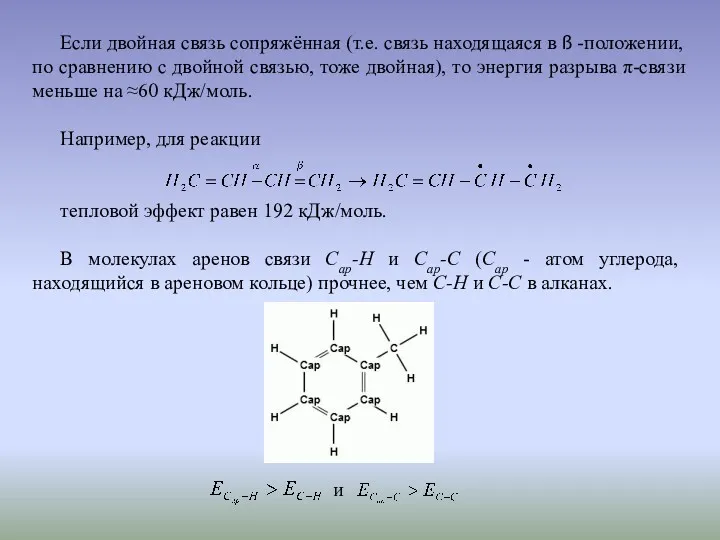
Если двойная связь сопряжённая (т.е. связь находящаяся в ß -положении, по
Если двойная связь сопряжённая (т.е. связь находящаяся в ß -положении, по

Прочность связи С-Н в циклоалкановых (нафтеновых) кольцах такая же, как связь
Прочность связи С-Н в циклоалкановых (нафтеновых) кольцах такая же, как связь

Свойства радикалов (продолжение)
4) Энергия связи С-Н у первого атома углерода в
Свойства радикалов (продолжение)
4) Энергия связи С-Н у первого атома углерода в

Лекция 3. Реакции радикалов.
Замещение и присоединение
В силу своей природы радикалы
Лекция 3. Реакции радикалов.
Замещение и присоединение
В силу своей природы радикалы

При наличии в молекуле связей С-Н с различной прочностью, реакция радикала
При наличии в молекуле связей С-Н с различной прочностью, реакция радикала

Реакции типа
вследствие энергетических (высокой энергии активации) и стерических (малый стерический множитель,
Реакции типа
вследствие энергетических (высокой энергии активации) и стерических (малый стерический множитель,

При реакции
затрачивается 249 кДж/моль на разрыв π-связи и выделяется в
При реакции
затрачивается 249 кДж/моль на разрыв π-связи и выделяется в

Лекция 4. Реакции радикалов.
Распад, изомеризация, рекомбинация и диспропорционирование, цепные реакции
Лекция 4. Реакции радикалов.
Распад, изомеризация, рекомбинация и диспропорционирование, цепные реакции

Когда радикал может распасться по нескольким направлениям, то вероятность распада по
Когда радикал может распасться по нескольким направлениям, то вероятность распада по
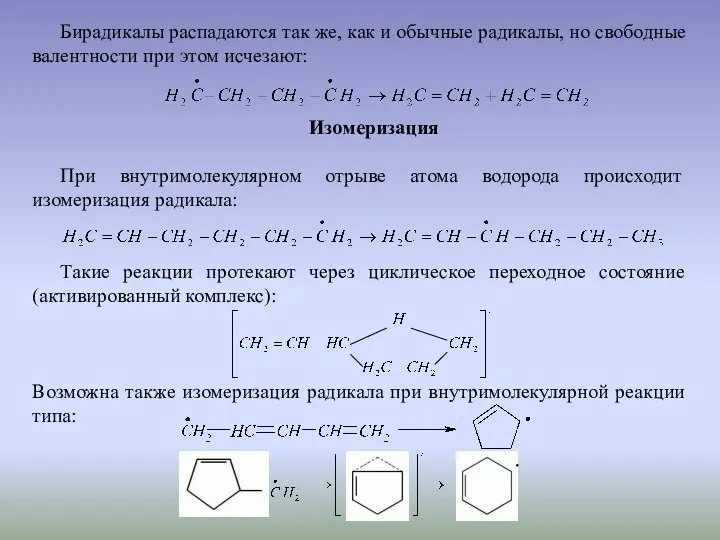
Бирадикалы распадаются так же, как и обычные радикалы, но свободные валентности
Бирадикалы распадаются так же, как и обычные радикалы, но свободные валентности

Алкановые радикалы не способны к 1,2-изомеризации. Однако для аренов наблюдается 1,2-перход,
Алкановые радикалы не способны к 1,2-изомеризации. Однако для аренов наблюдается 1,2-перход,

Цепные реакции свободных радикалов
Образовавшийся радикал при распаде или реакции с молекулой
Цепные реакции свободных радикалов
Образовавшийся радикал при распаде или реакции с молекулой

Суммарная реакция чередующихся элементарных реакций стадии звено цепи даёт уравнение распада
Суммарная реакция чередующихся элементарных реакций стадии звено цепи даёт уравнение распада

Лекция 5. Пиролиз в газовой фазе.
Термолиз алканов и алкенов
Пиролизом
Лекция 5. Пиролиз в газовой фазе.
Термолиз алканов и алкенов
Пиролизом

Термолиз алканов
Термолиз алканов приводит к образованию более термостабильных алканов и алкенов.
Термолиз алканов
Термолиз алканов приводит к образованию более термостабильных алканов и алкенов.

Помимо образующихся в больших количествах этилена и водорода (из 1 моль
Помимо образующихся в больших количествах этилена и водорода (из 1 моль

В процессе пиролиза н-бутана преобладают две реакции его распада:
1) ;
2) .
Нормальные
В процессе пиролиза н-бутана преобладают две реакции его распада:
1) ;
2) .
Нормальные

Самым термостабильным из гомологического ряда алкенов является этилен (деструкция начинается при
Самым термостабильным из гомологического ряда алкенов является этилен (деструкция начинается при

Пропилен по термической стабильности уступает этилену и при термолизе образует метан
Пропилен по термической стабильности уступает этилену и при термолизе образует метан

Лекция 6. Пиролиз в газовой фазе.
Термолиз нафтенов и ароматики
Термолиз
Лекция 6. Пиролиз в газовой фазе.
Термолиз нафтенов и ароматики
Термолиз

Дегидрирование незамещённых циклоалканов по цепному механизму не происходит, т.к. распад с
Дегидрирование незамещённых циклоалканов по цепному механизму не происходит, т.к. распад с

Механизм превращения циклогексана при термолизе:
Механизм превращения циклогексана при термолизе:

Механизм превращения циклопентана при термолизе:
Пироуглерод откладывается в трубах печи, ухудшая их
Механизм превращения циклопентана при термолизе:
Пироуглерод откладывается в трубах печи, ухудшая их

Термолиз ароматических углеводородов
Термоустойчивасть аренов зависит от наличия алкильных цепей в их
Термолиз ароматических углеводородов
Термоустойчивасть аренов зависит от наличия алкильных цепей в их

В результате конденсации нафталина образуется более высококонденсированная ароматика:
нафталин динафтил перилен
Ароматические углеводороды
В результате конденсации нафталина образуется более высококонденсированная ароматика:
нафталин динафтил перилен
Ароматические углеводороды

4) Алкены полимеризуются и вступают в реакции деструктивной полимеризации, также возможны
4) Алкены полимеризуются и вступают в реакции деструктивной полимеризации, также возможны

Лекция 7.
Жидкофазный термолиз
Жидкофазный термолиз имеет место в таких термодеструктивных
Лекция 7.
Жидкофазный термолиз
Жидкофазный термолиз имеет место в таких термодеструктивных

«Клеточный эффект»
Сущность «клеточного эффекта» заключается в том, что при распаде молекулы
«Клеточный эффект»
Сущность «клеточного эффекта» заключается в том, что при распаде молекулы

Анализ термического превращения углеводородов на примере процесса замедленного коксования
Одним из вариантов
Анализ термического превращения углеводородов на примере процесса замедленного коксования
Одним из вариантов

На каждой стадии образуются помимо продуктов уплотнения газы и более низкомолекулярные
На каждой стадии образуются помимо продуктов уплотнения газы и более низкомолекулярные

На скорость термодеструктивных превращений ТНО существенно влияет растворяющая способность дисперсионной среды
На скорость термодеструктивных превращений ТНО существенно влияет растворяющая способность дисперсионной среды
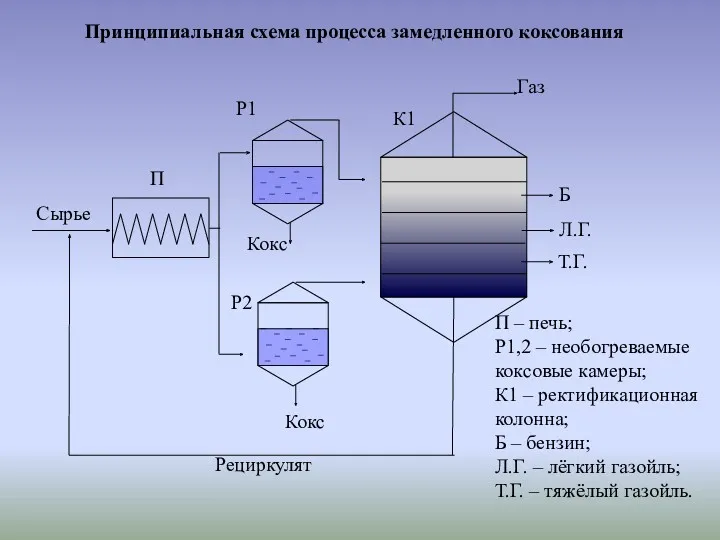
Принципиальная схема процесса замедленного коксования
Принципиальная схема процесса замедленного коксования

К основным закономерностям процесса коксования относятся
1. Сырье коксования должно быть с
К основным закономерностям процесса коксования относятся
1. Сырье коксования должно быть с

5. С повышением давления часть испарившихся углеводородов переходит в жидкую ДС,
5. С повышением давления часть испарившихся углеводородов переходит в жидкую ДС,

Лекция 8.
Образование и свойства карбоний-ионов
Как установлено выше, образование ионов
Лекция 8.
Образование и свойства карбоний-ионов
Как установлено выше, образование ионов

По природе промежуточного химического взаимодействия реагирующих химических веществ:
1) Гомолитический – когда
По природе промежуточного химического взаимодействия реагирующих химических веществ:
1) Гомолитический – когда

Энергия, необходимая для образования К-И, возрастает с увеличением числа атомов водорода,
Энергия, необходимая для образования К-И, возрастает с увеличением числа атомов водорода,

При этом скорее образуется вторичный К-И, нежели первичный, т.к. он более
При этом скорее образуется вторичный К-И, нежели первичный, т.к. он более

2) Неклассический способ
Кислоты бывают настолько сильными, что они способны протонировать даже
2) Неклассический способ
Кислоты бывают настолько сильными, что они способны протонировать даже

Таким образом, образование К-И по классическому способу происходит в соответствии с
Таким образом, образование К-И по классическому способу происходит в соответствии с

Лекция 9. Реакции карбоний-ионов.
Замещение и изомеризация
Замещение (перенос гидрид-иона)
Перенос гидрид-иона
Лекция 9. Реакции карбоний-ионов.
Замещение и изомеризация
Замещение (перенос гидрид-иона)
Перенос гидрид-иона

Бензин КК будет обладать большим октановым числом, так как содержание изопарафиновых
Бензин КК будет обладать большим октановым числом, так как содержание изопарафиновых

Перегруппировка (изомеризация)
Важной реакцией К-И является реакция перегруппировки путём сдвига атома водорода
Перегруппировка (изомеризация)
Важной реакцией К-И является реакция перегруппировки путём сдвига атома водорода

Изомеризация насыщенных углеводородов протекает через образование таких же К-И, но на
Изомеризация насыщенных углеводородов протекает через образование таких же К-И, но на

Лекция 10. Реакции карбоний-ионов.
Присоединение, распад и конденсация
Присоединение
К-И, как и
Лекция 10. Реакции карбоний-ионов.
Присоединение, распад и конденсация
Присоединение
К-И, как и
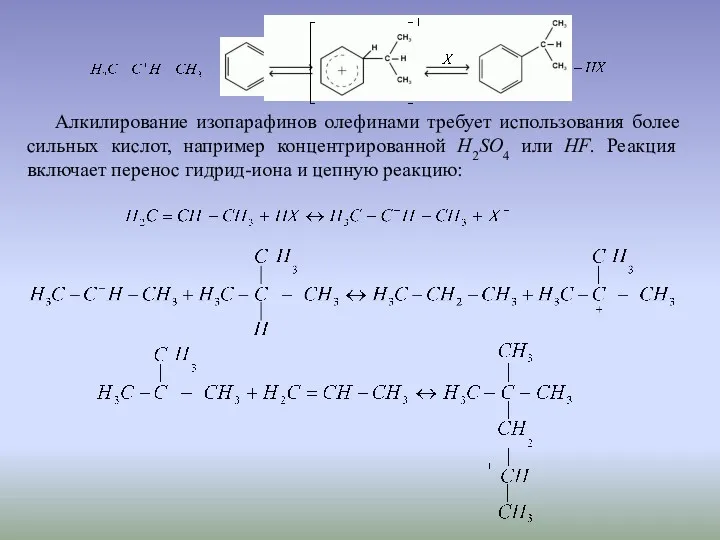
Алкилирование изопарафинов олефинами требует использования более сильных кислот, например концентрированной H2SO4
Алкилирование изопарафинов олефинами требует использования более сильных кислот, например концентрированной H2SO4

Распад (крекинг)
К-И, как и радикалы, с большой скоростью подвергаются реакции распада
Распад (крекинг)
К-И, как и радикалы, с большой скоростью подвергаются реакции распада

Продолжение крекинга прямой цепи по ß-связям приводит к образованию пропилена с
Продолжение крекинга прямой цепи по ß-связям приводит к образованию пропилена с
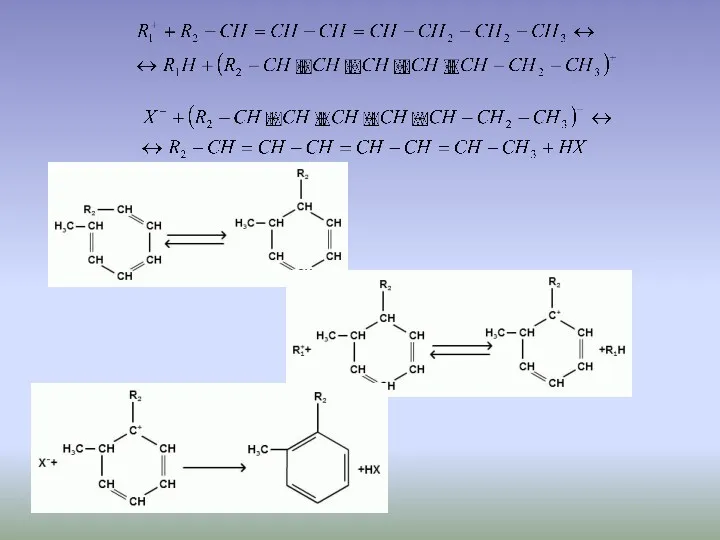
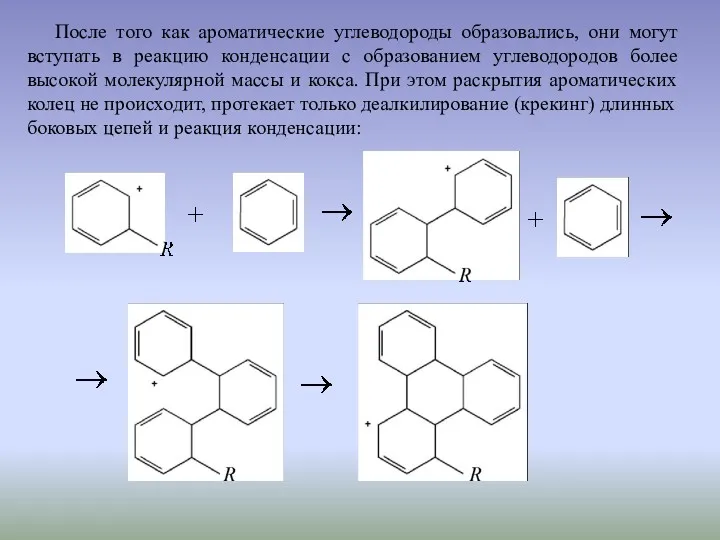
После того как ароматические углеводороды образовались, они могут вступать в реакцию
После того как ароматические углеводороды образовались, они могут вступать в реакцию

Лекция 11.
Характеристика катализа и катализаторов
Большинство промышленных процессов основано на
Лекция 11.
Характеристика катализа и катализаторов
Большинство промышленных процессов основано на
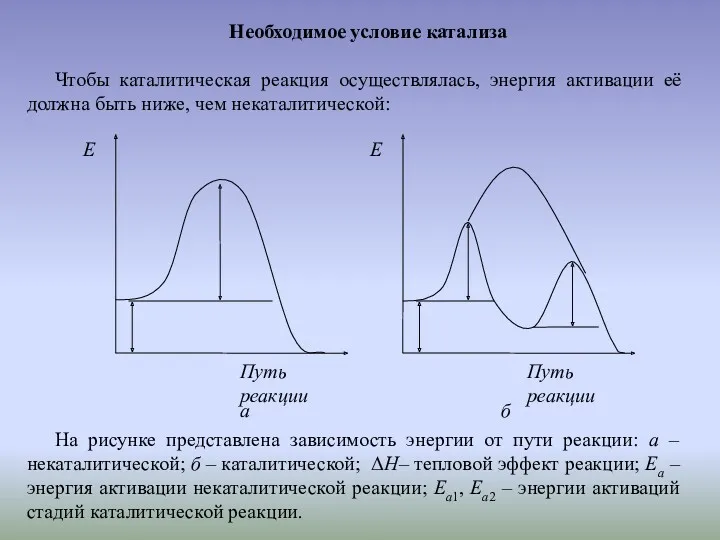
Необходимое условие катализа
Чтобы каталитическая реакция осуществлялась, энергия активации её должна быть
Необходимое условие катализа
Чтобы каталитическая реакция осуществлялась, энергия активации её должна быть

Активность и селективность катализатора
Скорость данной реакции в присутствии различных катализаторов характеризуется
Активность и селективность катализатора
Скорость данной реакции в присутствии различных катализаторов характеризуется

Стабильность катализатора
Стабильностью катализатора называется его способность сохранять активность во времени.
Жидкий катализатор
Стабильность катализатора
Стабильностью катализатора называется его способность сохранять активность во времени.
Жидкий катализатор

Химические изменения катализатора в процессе его работы вызываются хемосорбцией на его
Химические изменения катализатора в процессе его работы вызываются хемосорбцией на его

Кислотно-основный катализ
В кислотно-основных реакциях промежуточные активные частицы – ионы, и катализатор
Кислотно-основный катализ
В кислотно-основных реакциях промежуточные активные частицы – ионы, и катализатор

Окислительно-восстановительный катализ
В окислительно-восстановительных реакциях промежуточные частицы – радикалоподобные нейтральные образования, связанные
Окислительно-восстановительный катализ
В окислительно-восстановительных реакциях промежуточные частицы – радикалоподобные нейтральные образования, связанные

Координационно-комплексный (бифункциональный) катализ
В координационно-комплексном катализе при образовании комплекса реагента с катализатором
Координационно-комплексный (бифункциональный) катализ
В координационно-комплексном катализе при образовании комплекса реагента с катализатором

Лекция 12.
Алюмосиликатные катализаторы
Промышленное значение имеют катализаторы трёх типов:
Природные активированные
Лекция 12.
Алюмосиликатные катализаторы
Промышленное значение имеют катализаторы трёх типов:
Природные активированные

Катализаторы этого типа обладают слабой устойчивостью к действию высоких температур. Железо,
Катализаторы этого типа обладают слабой устойчивостью к действию высоких температур. Железо,

Протонные кислотные центры образуются, например, при замещении алюминием атомов кремния в
Протонные кислотные центры образуются, например, при замещении алюминием атомов кремния в

Апротонные кислотные центры могут иметь следующую структуру:
Атом алюминия в такой структуре
Апротонные кислотные центры могут иметь следующую структуру:
Атом алюминия в такой структуре
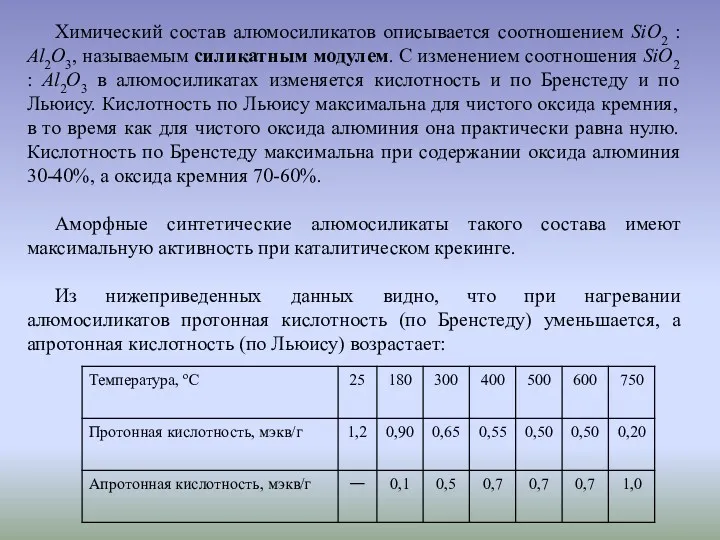
Химический состав алюмосиликатов описывается соотношением SiO2 : Al2O3, называемым силикатным модулем.
Химический состав алюмосиликатов описывается соотношением SiO2 : Al2O3, называемым силикатным модулем.
Синтетические кристаллические алюмосиликаты
Цеолиты (от греч. цео - кипящий, литос - камень)
Синтетические кристаллические алюмосиликаты
Цеолиты (от греч. цео - кипящий, литос - камень)
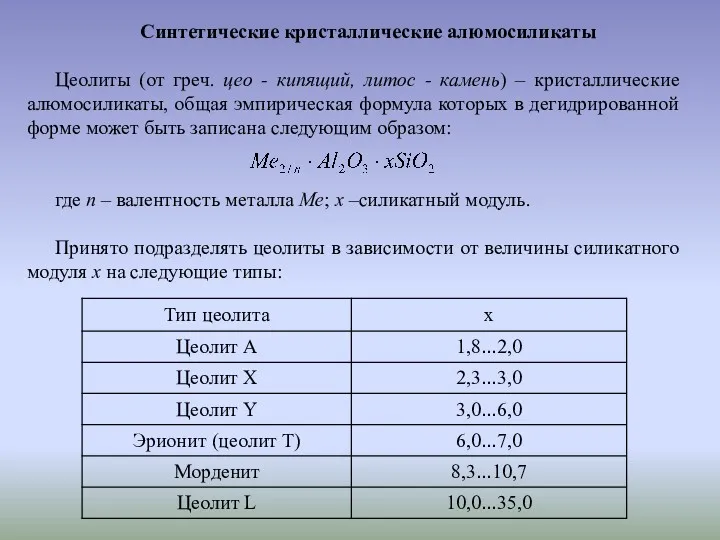
Цеолиты типа X и Y имеют кристаллическую структуру, образованную тетраэдрами SiO4
Цеолиты типа X и Y имеют кристаллическую структуру, образованную тетраэдрами SiO4
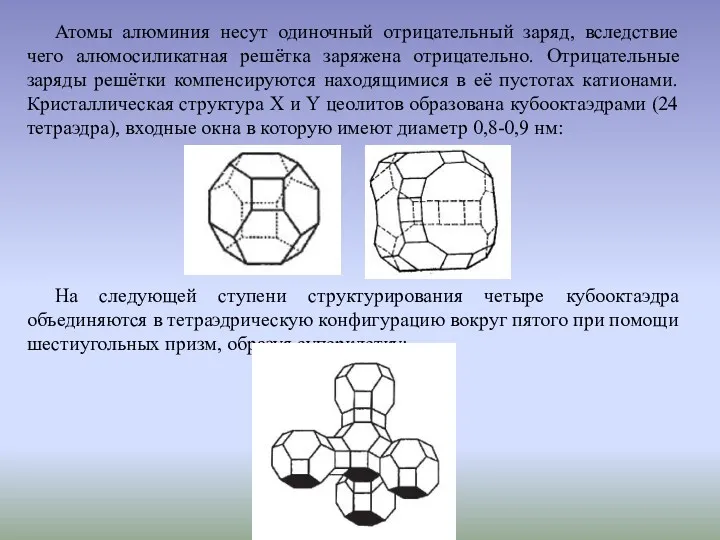
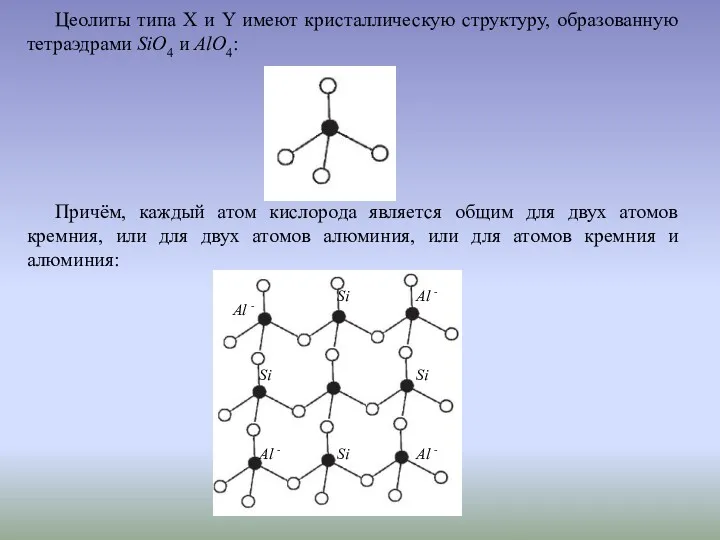
Атомы алюминия несут одиночный отрицательный заряд, вследствие чего алюмосиликатная решётка заряжена
Атомы алюминия несут одиночный отрицательный заряд, вследствие чего алюмосиликатная решётка заряжена
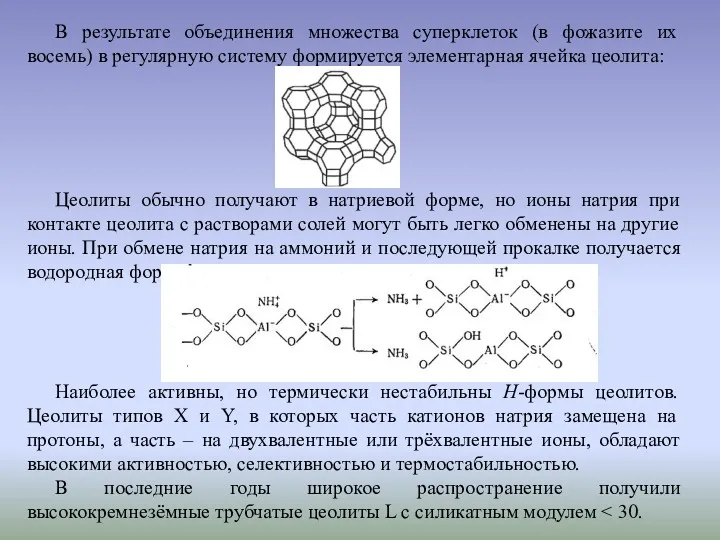
В результате объединения множества суперклеток (в фожазите их восемь) в регулярную
В результате объединения множества суперклеток (в фожазите их восемь) в регулярную

Промышленные катализаторы
Катализаторы современных крупнотоннажных процессов каталитического крекинга, осуществляемых при высоких температурах
Промышленные катализаторы
Катализаторы современных крупнотоннажных процессов каталитического крекинга, осуществляемых при высоких температурах

2) Активного компонента – цеолита;
На поверхности цеолитсодержащих катализаторов наблюдаются два типа
2) Активного компонента – цеолита;
На поверхности цеолитсодержащих катализаторов наблюдаются два типа

Перечень наиболее типичных вспомогательных добавок:
В качестве промотора, идентифицирующего регенерацию закоксованного катализатора,
Перечень наиболее типичных вспомогательных добавок:
В качестве промотора, идентифицирующего регенерацию закоксованного катализатора,

Лекция 13. Процессы, идущие по бифункциональному катализу: каталитический риформинг и изомеризация
Лекция 13. Процессы, идущие по бифункциональному катализу: каталитический риформинг и изомеризация
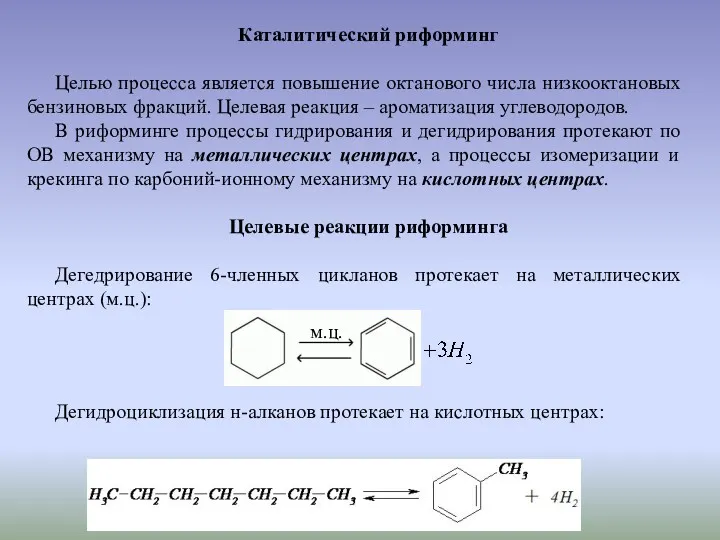
Каталитический риформинг
Целью процесса является повышение октанового числа низкооктановых бензиновых фракций. Целевая
Каталитический риформинг
Целью процесса является повышение октанового числа низкооктановых бензиновых фракций. Целевая
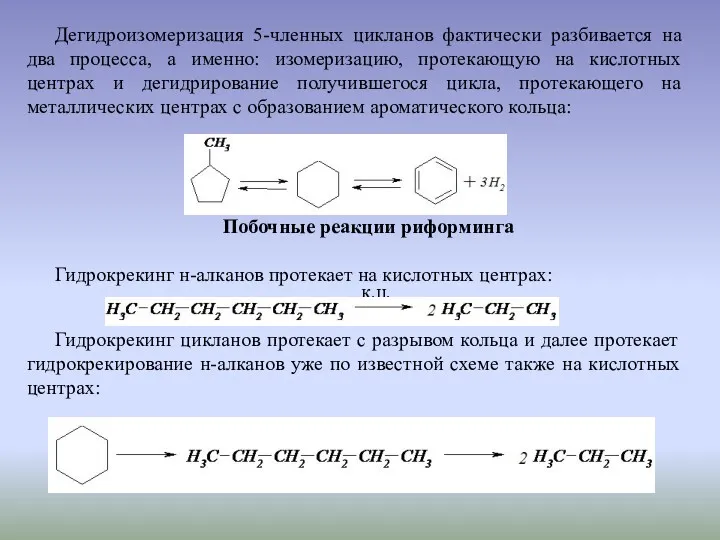
Дегидроизомеризация 5-членных цикланов фактически разбивается на два процесса, а именно: изомеризацию,
Дегидроизомеризация 5-членных цикланов фактически разбивается на два процесса, а именно: изомеризацию,
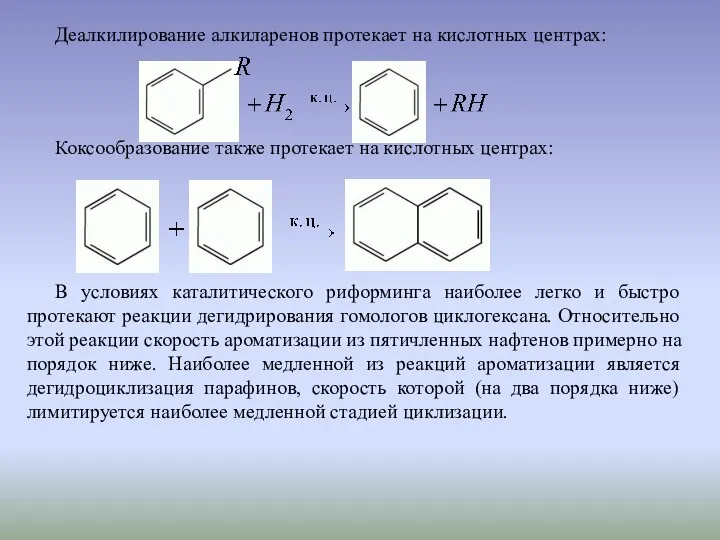
Деалкилирование алкиларенов протекает на кислотных центрах:
Коксообразование также протекает на кислотных центрах:
В
Деалкилирование алкиларенов протекает на кислотных центрах:
Коксообразование также протекает на кислотных центрах:
В

Изомеризация в бифункциональном катализе
На бифункциональных катализаторах, обладающих дегидро-гидрирующей и кислотной активностью,
Изомеризация в бифункциональном катализе
На бифункциональных катализаторах, обладающих дегидро-гидрирующей и кислотной активностью,

Давление не оказывает существенного влияния на стадию дегидрирования и гидрирования. Повышение
Давление не оказывает существенного влияния на стадию дегидрирования и гидрирования. Повышение

Лекция 14. Процессы, идущие по бифункциональному катализу: гидрокрекинг
Гидрокрекинг – каталитический
Лекция 14. Процессы, идущие по бифункциональному катализу: гидрокрекинг
Гидрокрекинг – каталитический
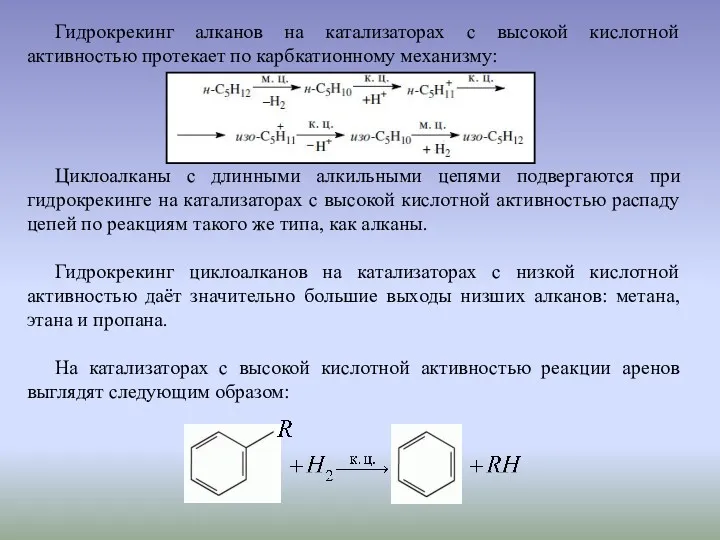
Гидрокрекинг алканов на катализаторах с высокой кислотной активностью протекает по карбкатионному
Гидрокрекинг алканов на катализаторах с высокой кислотной активностью протекает по карбкатионному
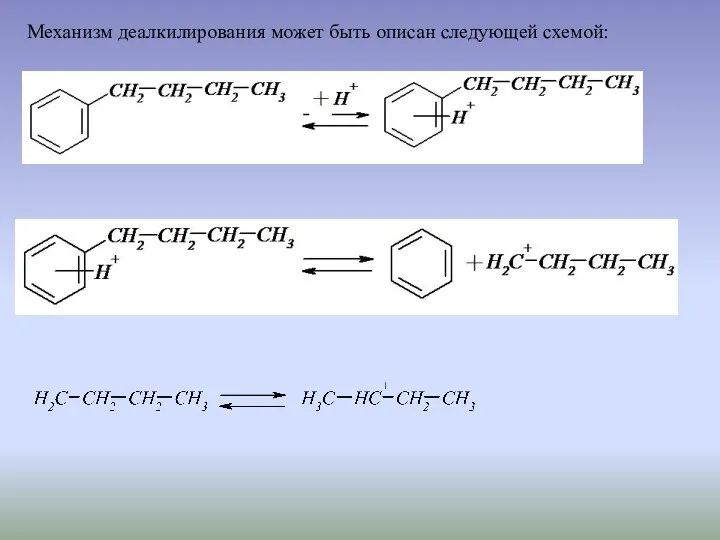
Механизм деалкилирования может быть описан следующей схемой:
Механизм деалкилирования может быть описан следующей схемой:

И далее возможны два пути протекания реакции. Первый с образованием в
И далее возможны два пути протекания реакции. Первый с образованием в

Лекция 15. Процессы, идущие по бифункциональному катализу: гидроочистка
Гидроочистка – процесс
Лекция 15. Процессы, идущие по бифункциональному катализу: гидроочистка
Гидроочистка – процесс

Гидроочистка серусодержащих соединений
Серусодержащие соединения, имеющиеся в нефтепродукте подвергаются следующим изменениям:
Меркаптаны гидрируются
Гидроочистка серусодержащих соединений
Серусодержащие соединения, имеющиеся в нефтепродукте подвергаются следующим изменениям:
Меркаптаны гидрируются

Тетрагидротиофены гидрируются с образованием соответствующих алифатических углеводородов:
Тиофены дают такие же продукты,
Тетрагидротиофены гидрируются с образованием соответствующих алифатических углеводородов:
Тиофены дают такие же продукты,
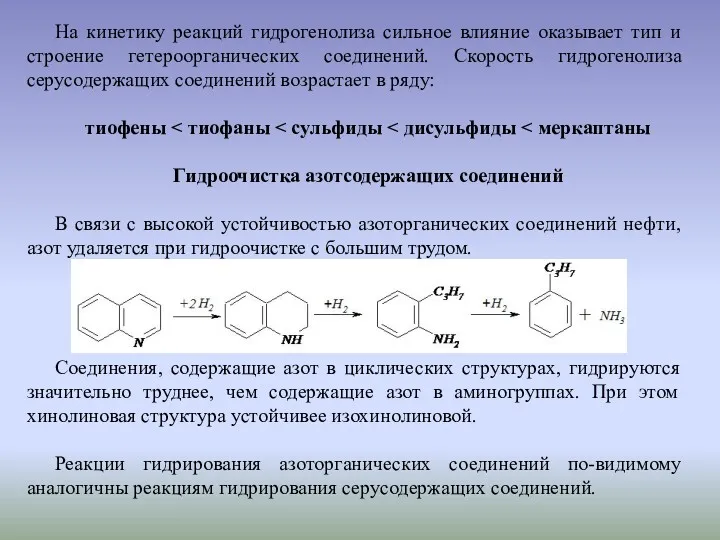
На кинетику реакций гидрогенолиза сильное влияние оказывает тип и строение гетероорганических
На кинетику реакций гидрогенолиза сильное влияние оказывает тип и строение гетероорганических
 Химическое равновесие. Принцип Ле Шателье
Химическое равновесие. Принцип Ле Шателье Соединения галогенов
Соединения галогенов Аминокислоты. Пептиды. Хроматографические методы исследования
Аминокислоты. Пептиды. Хроматографические методы исследования Химическая связь
Химическая связь Массовая доля вещества в растворе
Массовая доля вещества в растворе Визначення іонів лужних і лужноземельних іонів у природних водах
Визначення іонів лужних і лужноземельних іонів у природних водах Уникальная соль (для дошколников)
Уникальная соль (для дошколников) Пищевые добавки
Пищевые добавки Коллигативные свойства растворов
Коллигативные свойства растворов Основы термической и химико-термической обработки стали. Теория и технология термической обработки стали. Лекция 3. Тема 7
Основы термической и химико-термической обработки стали. Теория и технология термической обработки стали. Лекция 3. Тема 7 Литий. Физические свойства лития
Литий. Физические свойства лития Типы заданий. ЕГЭ №32
Типы заданий. ЕГЭ №32 20230419_izomery
20230419_izomery Растворы и растворители
Растворы и растворители Фосфор и его соединения
Фосфор и его соединения Общая характеристика неметаллов
Общая характеристика неметаллов Простые вещества. Игра Счастливый случай
Простые вещества. Игра Счастливый случай Элемент, имеющий относительную атомную массу
Элемент, имеющий относительную атомную массу Введение в аналитическую химию. Введение в качественный анализ
Введение в аналитическую химию. Введение в качественный анализ Алканы. Гомологи
Алканы. Гомологи Методы разделения и исследования состава нефти и газа
Методы разделения и исследования состава нефти и газа Формальная кинетика. Предмет химической кинетики
Формальная кинетика. Предмет химической кинетики Хром. Элемент под № 24
Хром. Элемент под № 24 Аммиак. Состав вещества
Аммиак. Состав вещества Галогены. Фтор, хлор, бром, йод, астат
Галогены. Фтор, хлор, бром, йод, астат Породообразующие минералы
Породообразующие минералы Особенности строения, реакционной способности и методы синтеза гидроксилсодержащих соединений
Особенности строения, реакционной способности и методы синтеза гидроксилсодержащих соединений Взрывоопасные грузы
Взрывоопасные грузы