- Формальная кинетика. Предмет химической кинетики

Содержание
- 2. Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем, что в термодинамике учитывается только
- 3. Можно выделить две главные задачи химической кинетики, определяющие ее практическое и теоретическое значение: экспериментальное исследование скорости
- 8. В связи с этим для элементарных реакций вводится понятие молекулярность – число молекул, принимающих участие в
- 11. Простые необратимые реакции В системе одновременно и независимо могут протекать реакции с различной скоростью, но в
- 14. Рис. 1. Зависимость концентраций прореагировавшего и оставшегося вещества от времени
- 26. Методы определения порядка реакции При определении порядка реакции вначале находят порядок по каждому из реагирующих веществ.
- 27. Метод графического подбора. Как следует из уравнения (10) для реакции первого порядка выполняется линейная зависимость в
- 28. 2. Метод аналитического подбора уравнения заключается в том, что проводится расчет константы скорости путем подстановки экспериментальных
- 29. Рис. 3. Определение порядка по периоду полуреакции
- 30. 4. Графический метод определения порядка. Скорость реакции n-ого порядка по данному веществу равна v = kcn
- 31. Рис. 4. Определение порядка реакции графическим методом: a) нахождение скорости; б) нахождение порядка и константы скорости
- 44. Рис. 5. Зависимость концентрации веществ от времени в последовательных реакциях
- 48. Метод стационарных концентраций В рассмотренном выше простейшем случае двух последовательных реакций первого порядка получены уравнения для
- 53. В заключение заметим, что метод стационарных концентраций не является совершенно строгим, его применение ограничивается выполнением условий
- 60. Как видно из уравнения (97), логарифм константы скорости является линейной функцией обратной температуры. Поэтому для экспериментального
- 61. Рис. 6. Зависимость lnk от 1/T
- 63. Рис. 7. Зависимость lnk от 1/T для параллельных реакций
- 69. Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED Так как энергия активации диффузии невелика, то
- 71. Скачать презентацию

Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем,
Такое кажущееся противоречие между теоретическими предсказаниями и практическими результатами обусловлено тем,

Можно выделить две главные задачи химической кинетики, определяющие ее практическое и
Можно выделить две главные задачи химической кинетики, определяющие ее практическое и





В связи с этим для элементарных реакций вводится понятие молекулярность –
В связи с этим для элементарных реакций вводится понятие молекулярность –



Простые необратимые реакции
В системе одновременно и независимо могут протекать реакции с
Простые необратимые реакции
В системе одновременно и независимо могут протекать реакции с


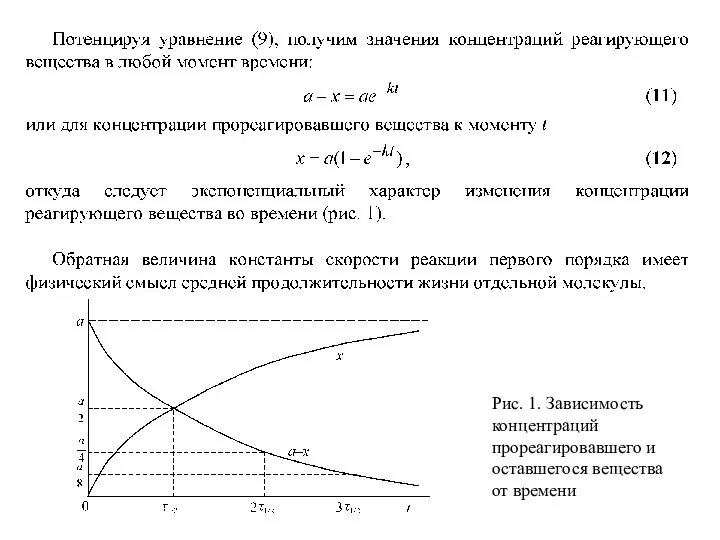
Рис. 1. Зависимость концентраций
прореагировавшего и оставшегося вещества
от времени
Рис. 1. Зависимость концентраций
прореагировавшего и оставшегося вещества
от времени












Методы определения порядка реакции
При определении порядка реакции вначале находят порядок по
Методы определения порядка реакции
При определении порядка реакции вначале находят порядок по

Метод графического подбора.
Как следует из уравнения (10) для реакции первого
Метод графического подбора.
Как следует из уравнения (10) для реакции первого

2. Метод аналитического подбора уравнения заключается в том, что проводится расчет
2. Метод аналитического подбора уравнения заключается в том, что проводится расчет

Рис. 3. Определение порядка
по периоду полуреакции
Рис. 3. Определение порядка
по периоду полуреакции

4. Графический метод определения порядка.
Скорость реакции n-ого порядка по данному
4. Графический метод определения порядка.
Скорость реакции n-ого порядка по данному
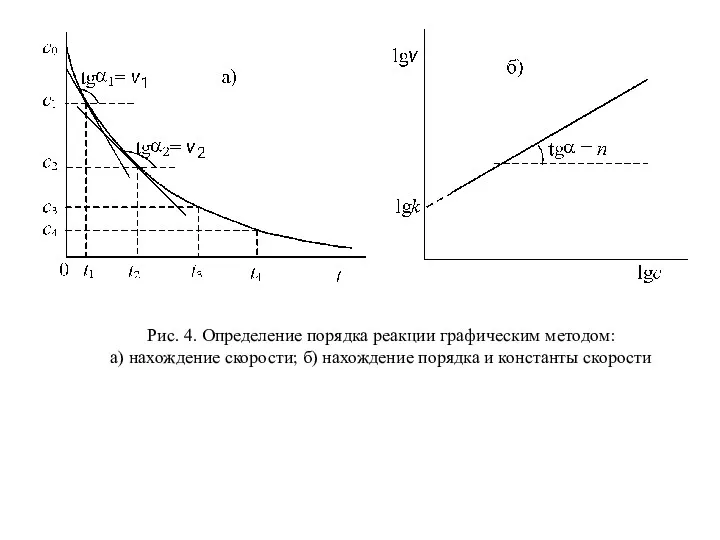
Рис. 4. Определение порядка реакции графическим методом:
a) нахождение скорости; б) нахождение
Рис. 4. Определение порядка реакции графическим методом:
a) нахождение скорости; б) нахождение












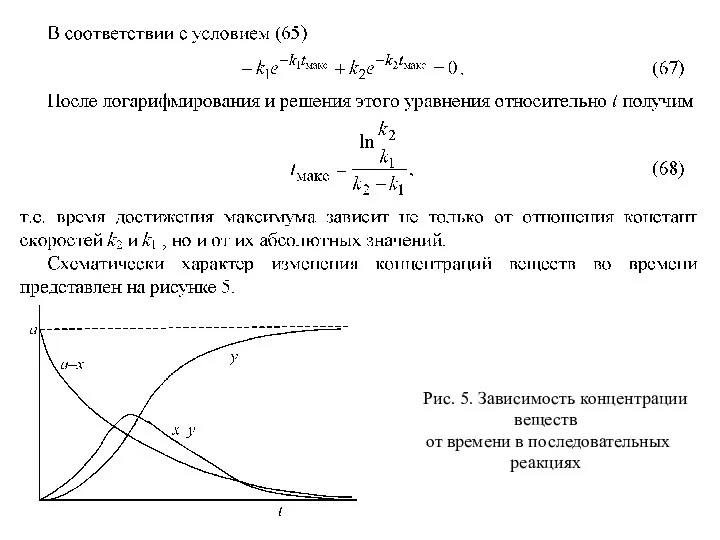
Рис. 5. Зависимость концентрации веществ
от времени в последовательных реакциях
Рис. 5. Зависимость концентрации веществ
от времени в последовательных реакциях




Метод стационарных концентраций
В рассмотренном выше простейшем случае двух последовательных реакций первого
Метод стационарных концентраций
В рассмотренном выше простейшем случае двух последовательных реакций первого





В заключение заметим, что метод стационарных концентраций не является совершенно строгим,
В заключение заметим, что метод стационарных концентраций не является совершенно строгим,







Как видно из уравнения (97), логарифм константы скорости является линейной функцией
Как видно из уравнения (97), логарифм константы скорости является линейной функцией
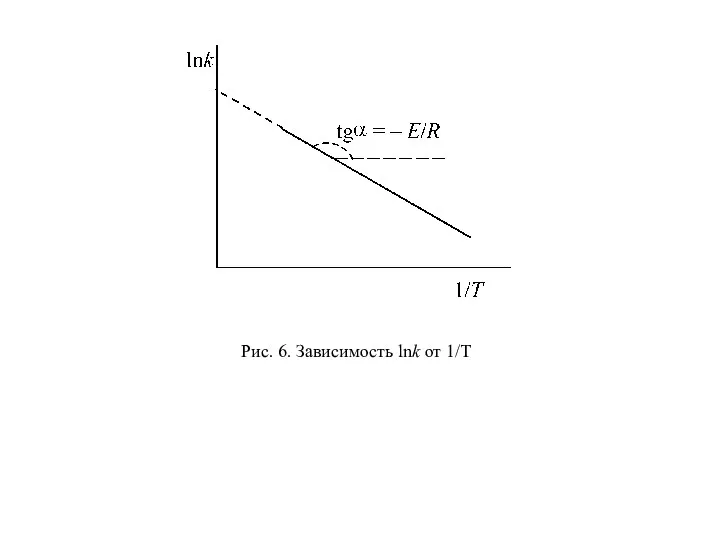
Рис. 6. Зависимость lnk от 1/T
Рис. 6. Зависимость lnk от 1/T

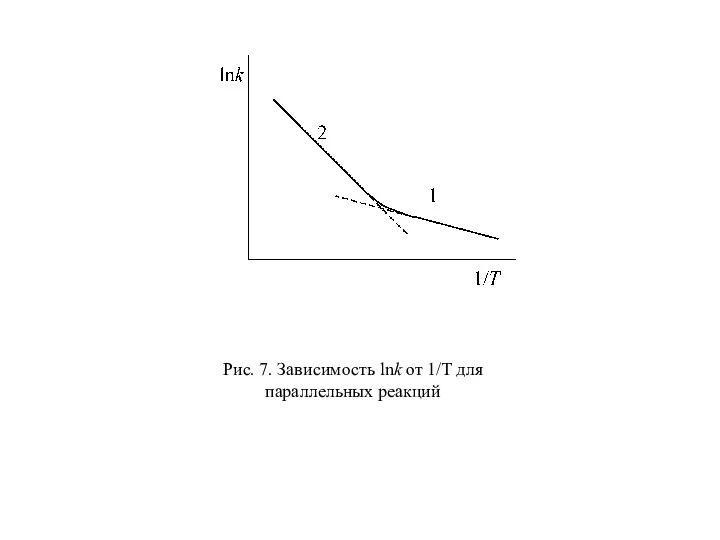
Рис. 7. Зависимость lnk от 1/T для параллельных реакций
Рис. 7. Зависимость lnk от 1/T для параллельных реакций






Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED << EA
Величина ED небольшая (5 ÷ 10 кДж/моль), т.е. ED << EA
 Вода
Вода Строение атома
Строение атома Железо и его соединения
Железо и его соединения Особенности строения соединений органической химии. 10 класс
Особенности строения соединений органической химии. 10 класс Історія відкриття періодичної системи хімічних елементів
Історія відкриття періодичної системи хімічних елементів Основы химмотологии моторных топлив. Тема 3
Основы химмотологии моторных топлив. Тема 3 Prezentatciya
Prezentatciya Растворы. Основные понятия. Концентрация. Законы Рауля
Растворы. Основные понятия. Концентрация. Законы Рауля Кислородсодержащие соединения азота
Кислородсодержащие соединения азота Аналитическая химия стойких органических загрязнителей
Аналитическая химия стойких органических загрязнителей Получение каталитического слоя на основе углеродных нанотрубок с наночастицами платины для водородно–воздушных топливных элементов
Получение каталитического слоя на основе углеродных нанотрубок с наночастицами платины для водородно–воздушных топливных элементов Периодическая система химических элементов Д.И. Менделеева. Строение атома
Периодическая система химических элементов Д.И. Менделеева. Строение атома Окисно-відновні реакції, їхнє значення. Складання найпростіших окисно-відновних реакцій, добір коефіцієнтів
Окисно-відновні реакції, їхнє значення. Складання найпростіших окисно-відновних реакцій, добір коефіцієнтів Химический элемент. Изотопы. 11 класс
Химический элемент. Изотопы. 11 класс Непредельные углеводороды
Непредельные углеводороды Аминокислоты. Пептиды. Хроматографические методы исследования
Аминокислоты. Пептиды. Хроматографические методы исследования Воздух и его состав
Воздух и его состав Поверхностный мембранный потенциал. Равновесие Доннана
Поверхностный мембранный потенциал. Равновесие Доннана Трансмиссионные масла
Трансмиссионные масла Одноатомные спирты. Глицерин
Одноатомные спирты. Глицерин Неметаллические материалы
Неметаллические материалы Углеводы. Общая характеристика углеводов
Углеводы. Общая характеристика углеводов Полимеры органические и неорганические
Полимеры органические и неорганические Первоначальные представления об органических веществах
Первоначальные представления об органических веществах Металлы. Свойства металлов
Металлы. Свойства металлов Алканы. Получение, свойства и применение
Алканы. Получение, свойства и применение Общая характеристика металлов
Общая характеристика металлов Кислоты, их состав и названия
Кислоты, их состав и названия