- Процессы ацилирования

Содержание
- 2. C-ацилирование аренов по Фриделю-Крафтсу (синтез ароматических кетонов) Ацилирующие агенты: в основном, карбоновые кислоты, их ангидриды и
- 3. Отличие реакций ацилирования и алкилирования аренов по Фриделю-Крафтсу В отличие от алкилирования, процессы ацилирования аренов: а)
- 4. Механизм образования электрофильных частиц В результате взаимодействия ацилирующих агентов с катализатором образуются электрофильные частицы: - во-первых,
- 5. Количество кислоты Льюиса должно быть не менее 1 моль на моль субстрата, так как катализатор взаимодействует
- 6. С-ацилирование аренов хлорангидридами кислот Хлорангидриды кислот — самые активные ацилирующие агенты, но малодоступные, нестабильные, дорогие (легко
- 7. С-ацилирование аренов ангидридами кислот встречается значительно реже, хотя активные, устойчивые, мало токсичные и агрессивные ; недостаток
- 8. С-ацилирование карбоновыми кислотами встречается редко из-за малой активности реагента, который наиболее доступен, стабилен, дешев, наименее токсичный
- 9. Ацилирование аренов по Гаттерману-Коху (синтез альдегидов) Хлорангидрид муравьиной кислоты нестабильное соединение и в реакциях Фриделя-Крафтса не
- 10. Реакция Вильсмайера (синтез ароматических альдегидов) Реагент обычно диметилформамид (ацильное соединение). Катализатор - хлорокись фосфора. Электрофильная частица
- 11. Реакция Реймера-Тимана (синтез ароматических гидроксиальдегидов) Субстратом являются фенолы с заместителями первого рода, нафтолы и другие активные
- 12. Реакция Кольбе-Шмидта (синтез ароматических гидроксикислот) Субстратом являются фенолы и аминофенолы. Ацилирующий агент – ангидрид угольной кислоты.
- 13. N-ацилирование (синтез амидов кислот) применяется как для получения нового соединения, так и для защиты аминогруппы; ацилирующие
- 14. Реакционная способность ацильных соединений Определяется величиной положительного заряда на атоме углерода карбонильной группы и способностью уходящей
- 15. Влияние уходящей группы на δ+ ацильной группы В ацильных соединениях одной и той же кислоты, величина
- 16. Способность группы Y уходить чем более сильным основанием является Y , тем хуже уходит. При определении
- 17. Хлорангидриды карбоновых кислот (+) самые активные ацилирующие агенты; реакции необратимые; реагенты можно брать в стехиометрических соотношениях.
- 18. Ангидриды карбоновых кислот (+) активные ацилирующие агенты, реакции необратимы, используют стехиометрические соотношения реагентов. (-) дороже и
- 19. Карбоновые кислоты (+) наиболее дешевые и доступные; (-) значительно менее активные реагенты, чем их ангидриды, образуют
- 20. Формилирование и ацетилирование аминов проводят в избытке кислоты (с муравьиной кислотой при 150 °С, с уксусной
- 21. Сложные эфиры карбоновых кислот в большинстве своем малоактивны, но не образуют солей с аминами и реагируют
- 22. N-ацилирование амидами карбоновых кислот применяют очень редко из-за малой активности реагента. Тем не менее, известны реакции,
- 23. О-Ацилирование (синтез сложных эфиров) проводится реже, чем аминогрупп и идет менее энергично. Механизмы О- и N-ацилирования,
- 24. О-Ацилирование хлорангидридами кислот Для связывания выделяющегося хлористого водорода применяют основания или ведут реакцию в таких условиях
- 25. О-Ацилирование ангидридами кислот используется только половина молекулы, поэтому в промышленности применяют, в основном, уксусный и фталевый
- 26. О-Ацилирование карбоновыми кислотами наиболее дешевый и доступный реагент но значительно менее активный, чем ангидриды, не взаимодействует
- 27. Механизм реакции этерификации SNAE Схема включает: активацию СООН, AN, создания хорошо уходящей группы и E уходящей
- 29. Скачать презентацию

C-ацилирование аренов по Фриделю-Крафтсу (синтез ароматических кетонов)
Ацилирующие агенты: в основном, карбоновые
C-ацилирование аренов по Фриделю-Крафтсу (синтез ароматических кетонов)
Ацилирующие агенты: в основном, карбоновые
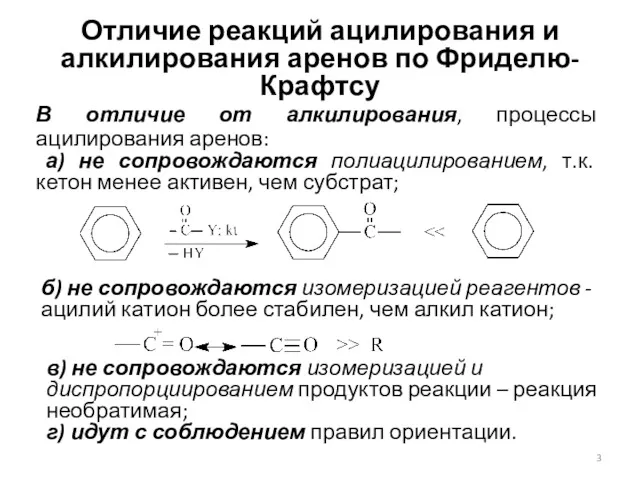
Отличие реакций ацилирования и алкилирования аренов по Фриделю-Крафтсу
В отличие от алкилирования,
Отличие реакций ацилирования и алкилирования аренов по Фриделю-Крафтсу
В отличие от алкилирования,

Механизм образования электрофильных частиц
В результате взаимодействия ацилирующих агентов с катализатором образуются
Механизм образования электрофильных частиц
В результате взаимодействия ацилирующих агентов с катализатором образуются

Количество кислоты Льюиса
должно быть не менее 1 моль на моль
Количество кислоты Льюиса
должно быть не менее 1 моль на моль

С-ацилирование аренов хлорангидридами кислот
Хлорангидриды кислот — самые активные ацилирующие агенты, но
С-ацилирование аренов хлорангидридами кислот
Хлорангидриды кислот — самые активные ацилирующие агенты, но
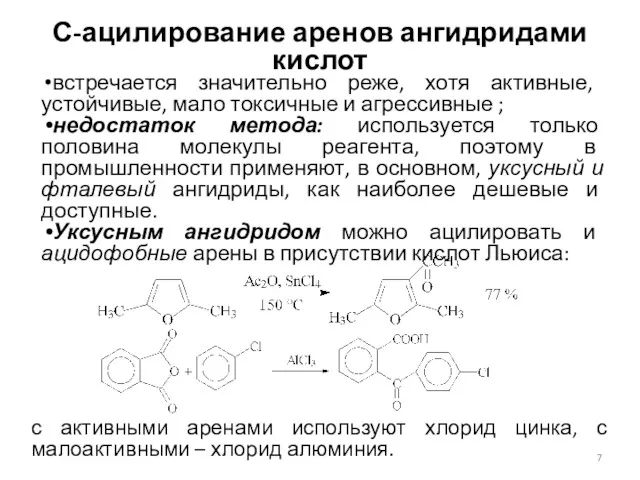
С-ацилирование аренов ангидридами кислот
встречается значительно реже, хотя активные, устойчивые, мало токсичные
С-ацилирование аренов ангидридами кислот
встречается значительно реже, хотя активные, устойчивые, мало токсичные
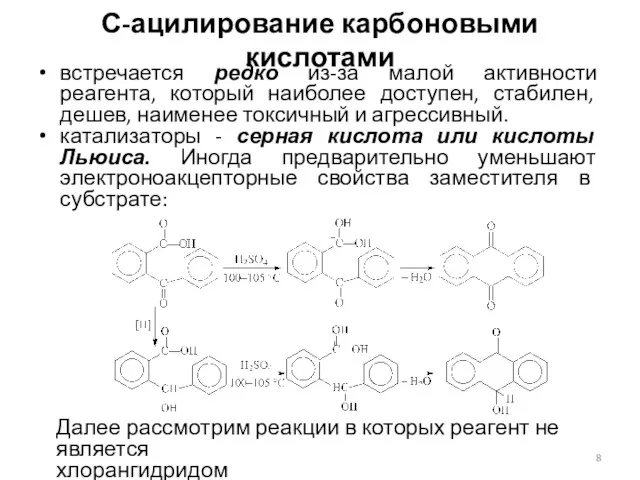
С-ацилирование карбоновыми кислотами
встречается редко из-за малой активности реагента, который наиболее доступен,
С-ацилирование карбоновыми кислотами
встречается редко из-за малой активности реагента, который наиболее доступен,

Ацилирование аренов по Гаттерману-Коху (синтез альдегидов)
Хлорангидрид муравьиной кислоты нестабильное соединение и
Ацилирование аренов по Гаттерману-Коху (синтез альдегидов)
Хлорангидрид муравьиной кислоты нестабильное соединение и
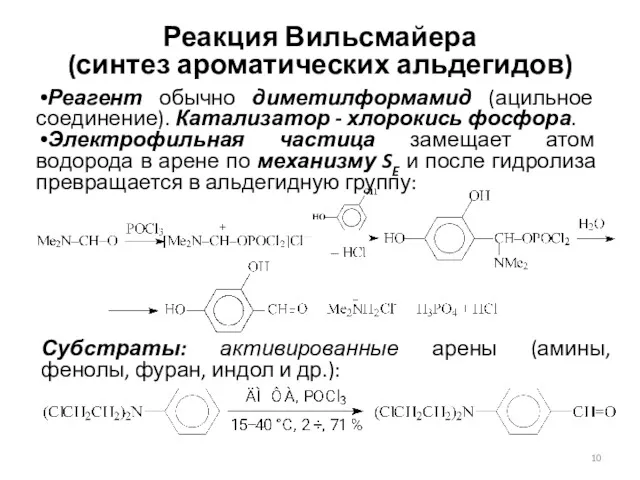
Реакция Вильсмайера
(синтез ароматических альдегидов)
Реагент обычно диметилформамид (ацильное соединение). Катализатор -
Реакция Вильсмайера
(синтез ароматических альдегидов)
Реагент обычно диметилформамид (ацильное соединение). Катализатор -
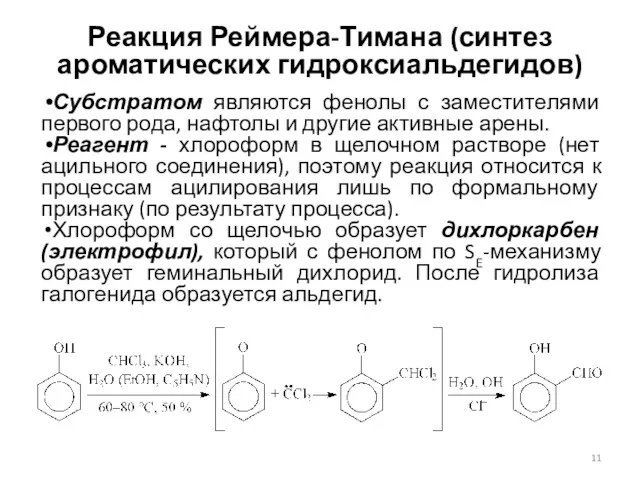
Реакция Реймера-Тимана (синтез ароматических гидроксиальдегидов)
Субстратом являются фенолы с заместителями первого рода,
Реакция Реймера-Тимана (синтез ароматических гидроксиальдегидов)
Субстратом являются фенолы с заместителями первого рода,
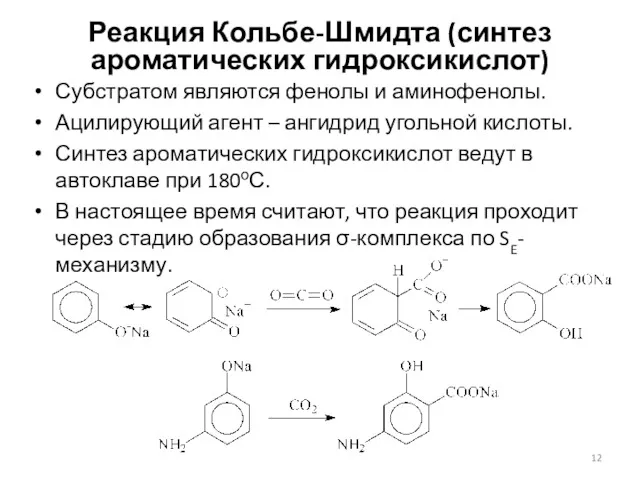
Реакция Кольбе-Шмидта (синтез ароматических гидроксикислот)
Субстратом являются фенолы и аминофенолы.
Ацилирующий агент
Реакция Кольбе-Шмидта (синтез ароматических гидроксикислот)
Субстратом являются фенолы и аминофенолы.
Ацилирующий агент

N-ацилирование (синтез амидов кислот)
применяется как для получения нового соединения, так и
N-ацилирование (синтез амидов кислот)
применяется как для получения нового соединения, так и

Реакционная способность ацильных соединений
Определяется величиной положительного заряда на атоме углерода карбонильной
Реакционная способность ацильных соединений
Определяется величиной положительного заряда на атоме углерода карбонильной
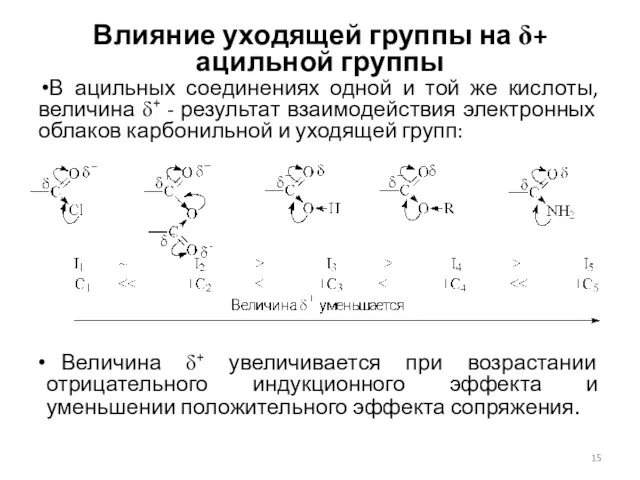
Влияние уходящей группы на δ+ ацильной группы
В ацильных соединениях одной и
Влияние уходящей группы на δ+ ацильной группы
В ацильных соединениях одной и

Способность группы Y уходить
чем более сильным основанием является Y , тем
Способность группы Y уходить
чем более сильным основанием является Y , тем
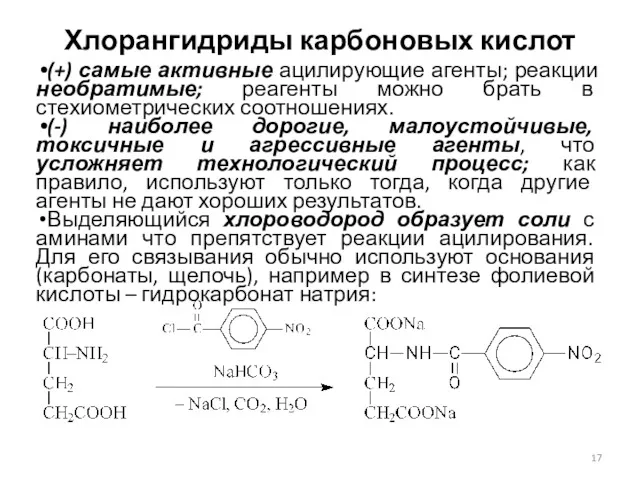
Хлорангидриды карбоновых кислот
(+) самые активные ацилирующие агенты; реакции необратимые; реагенты можно
Хлорангидриды карбоновых кислот
(+) самые активные ацилирующие агенты; реакции необратимые; реагенты можно
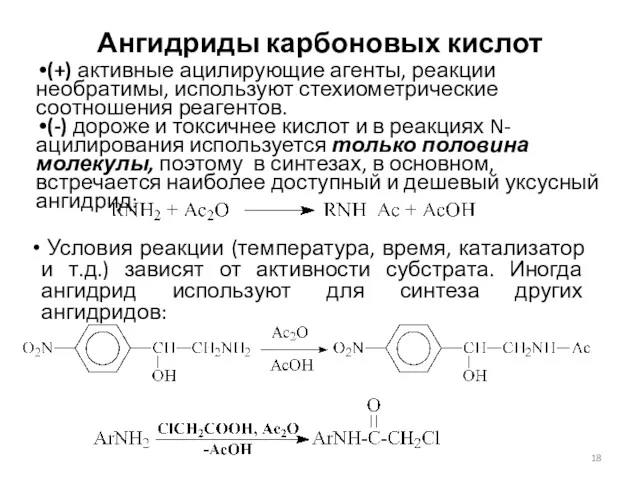
Ангидриды карбоновых кислот
(+) активные ацилирующие агенты, реакции необратимы, используют стехиометрические соотношения
Ангидриды карбоновых кислот
(+) активные ацилирующие агенты, реакции необратимы, используют стехиометрические соотношения

Карбоновые кислоты
(+) наиболее дешевые и доступные;
(-) значительно менее активные
Карбоновые кислоты
(+) наиболее дешевые и доступные;
(-) значительно менее активные
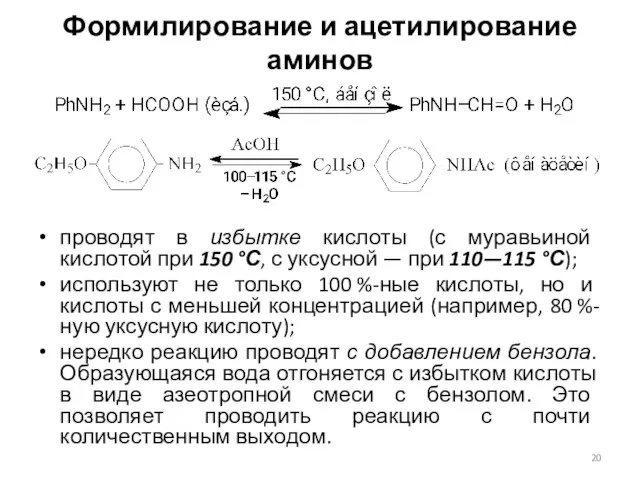
Формилирование и ацетилирование аминов
проводят в избытке кислоты (с муравьиной кислотой
Формилирование и ацетилирование аминов
проводят в избытке кислоты (с муравьиной кислотой
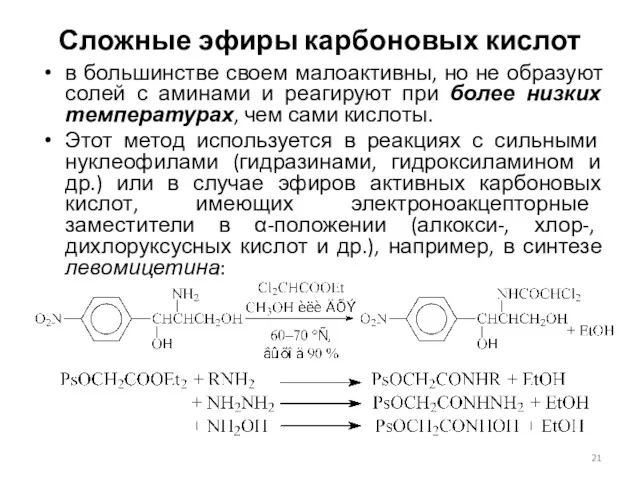
Сложные эфиры карбоновых кислот
в большинстве своем малоактивны, но не образуют солей
Сложные эфиры карбоновых кислот
в большинстве своем малоактивны, но не образуют солей

N-ацилирование амидами карбоновых кислот
применяют очень редко из-за малой активности реагента. Тем
N-ацилирование амидами карбоновых кислот
применяют очень редко из-за малой активности реагента. Тем
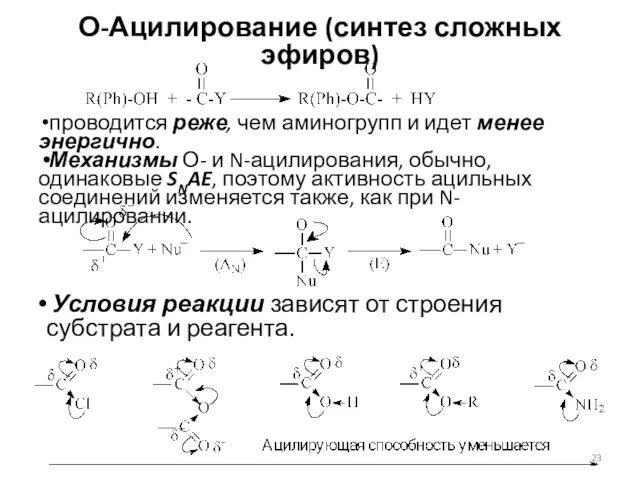
О-Ацилирование (синтез сложных эфиров)
проводится реже, чем аминогрупп и идет менее энергично.
О-Ацилирование (синтез сложных эфиров)
проводится реже, чем аминогрупп и идет менее энергично.

О-Ацилирование хлорангидридами кислот
Для связывания выделяющегося хлористого водорода применяют основания или ведут
О-Ацилирование хлорангидридами кислот
Для связывания выделяющегося хлористого водорода применяют основания или ведут
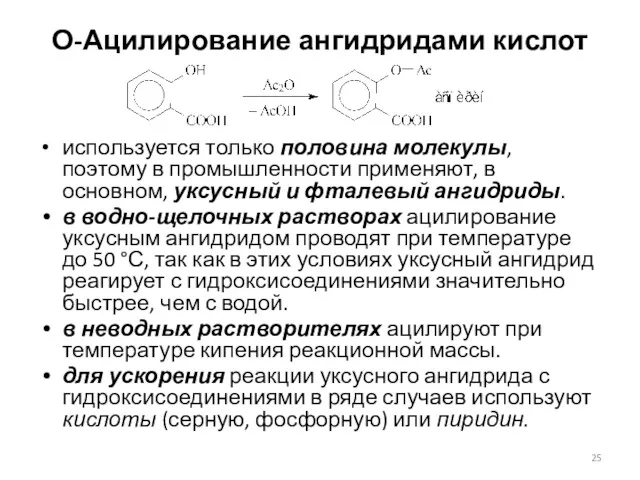
О-Ацилирование ангидридами кислот
используется только половина молекулы, поэтому в промышленности применяют, в
О-Ацилирование ангидридами кислот
используется только половина молекулы, поэтому в промышленности применяют, в

О-Ацилирование карбоновыми кислотами
наиболее дешевый и доступный реагент
но значительно менее активный,
О-Ацилирование карбоновыми кислотами
наиболее дешевый и доступный реагент
но значительно менее активный,
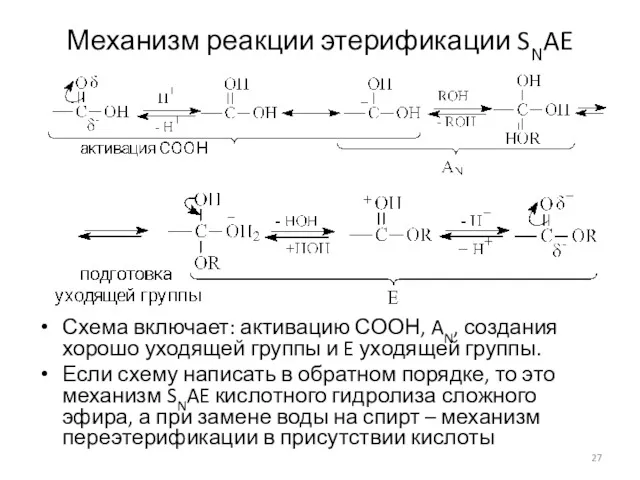
Механизм реакции этерификации SNAE
Схема включает: активацию СООН, AN, создания хорошо уходящей
Механизм реакции этерификации SNAE
Схема включает: активацию СООН, AN, создания хорошо уходящей
 Обобщение и систематизация по теме Соединения химических элементов. 8 класс
Обобщение и систематизация по теме Соединения химических элементов. 8 класс Периодическая система химических элементов. Периоды
Периодическая система химических элементов. Периоды Химическая кинетика. Часть II. Скорость химической реакции - развитие реакции во времени
Химическая кинетика. Часть II. Скорость химической реакции - развитие реакции во времени Энергетикалық деңгейлер
Энергетикалық деңгейлер Типы кристаллических решеток. Повторение: виды химической связи
Типы кристаллических решеток. Повторение: виды химической связи Минерал турмалин
Минерал турмалин Изомеры – это вещества, имеющие одинаковый состав
Изомеры – это вещества, имеющие одинаковый состав Массасы 4,2 г көміртек (IV) оксиді сумен әрекеттескенде қанша грамм көмір қышқылы (Н2СО3) түзілетінін есепте
Массасы 4,2 г көміртек (IV) оксиді сумен әрекеттескенде қанша грамм көмір қышқылы (Н2СО3) түзілетінін есепте Горение топлива
Горение топлива Химические свойства альдегидов
Химические свойства альдегидов Молярный объём газов
Молярный объём газов Анализ начальных участков изотерм адсорбции
Анализ начальных участков изотерм адсорбции Цинк
Цинк Серная кислота. ОХТ, лекция №7
Серная кислота. ОХТ, лекция №7 20230816_himiya_spirty
20230816_himiya_spirty Хімічні властивості кисню
Хімічні властивості кисню Периодический закон Д.И. Менделеева. Историческая формулировка периодического закона
Периодический закон Д.И. Менделеева. Историческая формулировка периодического закона Типы химических реакций
Типы химических реакций Амины, аминокислоты; состав, получение, значение, применение
Амины, аминокислоты; состав, получение, значение, применение Нанотехнологии. Отдельные представители наночастиц
Нанотехнологии. Отдельные представители наночастиц Классификация веществ
Классификация веществ Хром. Элемент под № 24
Хром. Элемент под № 24 Оксиды. 9 класс
Оксиды. 9 класс Основы химической кинетики
Основы химической кинетики Стереографическая проекция. Ориентация кристаллов высшей категории
Стереографическая проекция. Ориентация кристаллов высшей категории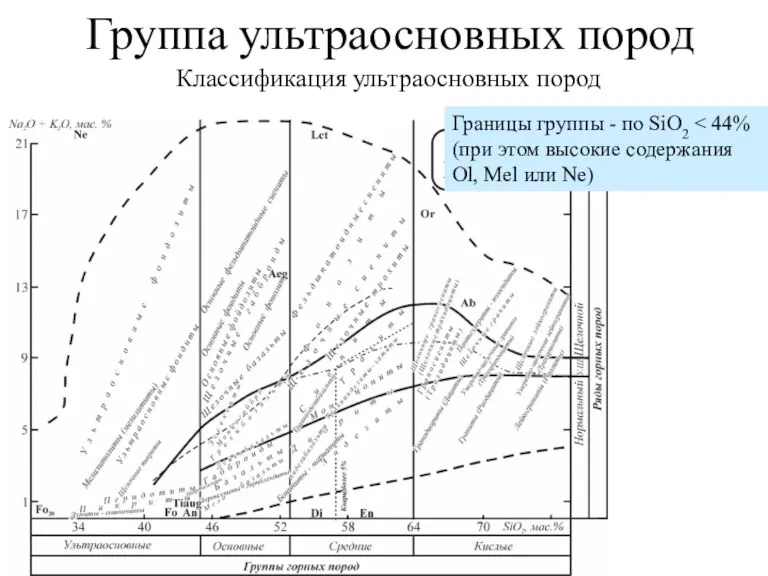 Группа ультраосновных пород
Группа ультраосновных пород Элементы химической термодинамики
Элементы химической термодинамики Этанол (эти́ловый спирт)
Этанол (эти́ловый спирт)