- Basis Sets and Pseudopotentials

Содержание
- 2. Slater-Type Orbitals (STO’s) N is a normalization constant a, b, and c determine the angular momentum,
- 3. Gaussian-Type Orbitals (GTO’s) N is a normalization constant a, b, and c determine the angular momentum,
- 4. Contracted Basis Sets P=primitive, C=contracted Reduces the number of basis functions The contraction coefficients, αi, are
- 5. Contracted Basis Sets Jensen, Figure 5.3, p. 202
- 6. STO-NG: STO approximated by linear combination of N Gaussians
- 7. Even-tempered Basis Sets Same functional form as the Gaussian functions used earlier The exponent, ζ, is
- 8. Well-tempered Basis Sets α, β, γ, and δ are parameters optimized to minimize the SCF energy
- 9. Davidson, E. R.; Feller, D. Chem. Rev. 1986, 86, 681-696.
- 10. Used to model infinite systems (e.g. metals, crystals, etc.) In infinite systems, molecular orbitals become bands
- 11. Polarization Functions Similar exponent as valence function Higher angular momentum (l+1) Uncontracted Gaussian (coefficient=1) Introduces flexibility
- 12. Diffuse Functions Smaller exponent than valence functions (larger spatial extent) Same angular momentum as valence functions
- 13. Cartesian vs. Spherical Cartesians: s – 1 function p – 3 functions d – 6 functions
- 14. Cartesian vs. Spherical Suppose we calculated the energy of HCl using a cc-pVDZ basis set using
- 15. Pople Basis Sets Optimized using Hartree-Fock Names have the form k-nlm++G** or k-nlmG(…) k is the
- 16. Pople Basis Sets Examples: 6-31G Three contracted Gaussians for the core with the valence represented by
- 17. Dunning Correlatoin Consistent Basis Sets Optimized using a correlated method (CIS, CISD, etc.) Names have the
- 18. Dunning Basis Sets Examples: cc-pVDZ Double zeta with polarization aug-cc-pVTZ Triple zeta with polarization and diffuse
- 19. Extrapolate to complete basis set limit Most useful for electron correlation methods P(lmax) = P(CBS) +
- 20. Basis Set Superposition Error Occurs when a basis function centered at one nucleus contributes the the
- 21. Counterpoise Correction E(A)ab is the energy of fragment A with the basis functions for A+B E(A)a
- 22. Additional Information EMSL Basis Set Exchange: https://bse.pnl.gov/bse/portal Further reading: Davidson, E. R.; Feller, D. Chem. Rev.
- 23. Effective Core Potentials (ECPs) and Model Core Potentials (MCPs)
- 24. Frozen Core Approximation Approximation made: atomic core orbitals are not allowed to change upon molecular formation;
- 25. Pseudopotentials - ECPs Effective core potentials (ECPs) are pseudopotentials that replace core electrons by a potential
- 26. Shape Consistent ECPs Nodeless pseudo-orbitals that resemble the valence orbitals in the bonding region The fit
- 27. Energy Consistent ECPs Approach that tries to reproduce the low-energy atomic spectrum (via correlated calculations) Usually
- 28. Pseudo-orbitals Visscher, L., “Relativisitic Electronic Structure Theory”, 2006 Winter School, Helkinki, Finland.
- 29. Large and Small Core ECPs Jensen, Figure 5.7, p. 224.
- 30. Pseudopotentials - MCPs Model Core Potentials (MCP) provide a computationally feasible treatment of heavy elements. MCPs
- 31. MCP Formulation All-electron (AE) Hamiltonian: MCP Hamiltonian: First term is the 1 electron MCP Hamiltonian Second
- 32. 1-electron Hamiltonian All-electron (AE) Hamiltonian: MCP Hamiltonian: First term is the 1 electron MCP Hamiltonian Second
- 33. MCP Nuclear Attraction AI, αI, BJ, and βJ are fitted MCP parameters MCP parameters are fitted
- 35. Скачать презентацию

Slater-Type Orbitals (STO’s)
N is a normalization constant
a, b, and
Slater-Type Orbitals (STO’s)
N is a normalization constant
a, b, and

Gaussian-Type Orbitals (GTO’s)
N is a normalization constant
a, b, and
Gaussian-Type Orbitals (GTO’s)
N is a normalization constant
a, b, and

Contracted Basis Sets
P=primitive, C=contracted
Reduces the number of basis functions
The
Contracted Basis Sets
P=primitive, C=contracted
Reduces the number of basis functions
The
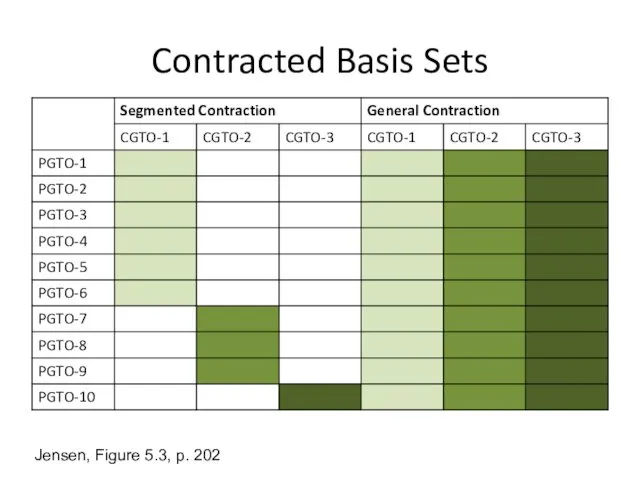
Contracted Basis Sets
Jensen, Figure 5.3, p. 202
Contracted Basis Sets
Jensen, Figure 5.3, p. 202
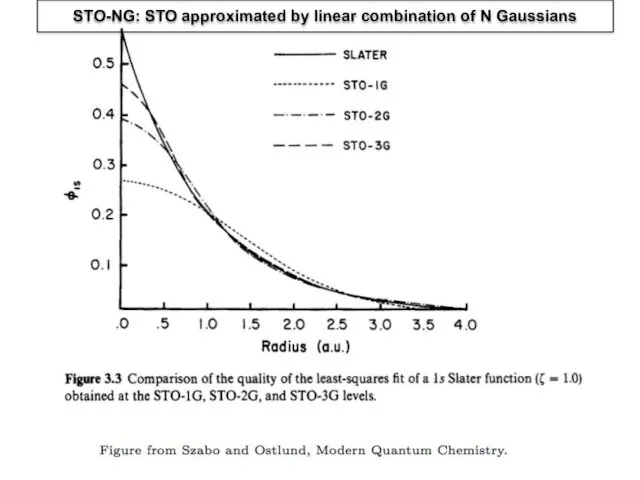
STO-NG: STO approximated by linear combination of N Gaussians
STO-NG: STO approximated by linear combination of N Gaussians

Even-tempered Basis Sets
Same functional form as the Gaussian functions used
Even-tempered Basis Sets
Same functional form as the Gaussian functions used

Well-tempered Basis Sets
α, β, γ, and δ are parameters optimized to
Well-tempered Basis Sets
α, β, γ, and δ are parameters optimized to
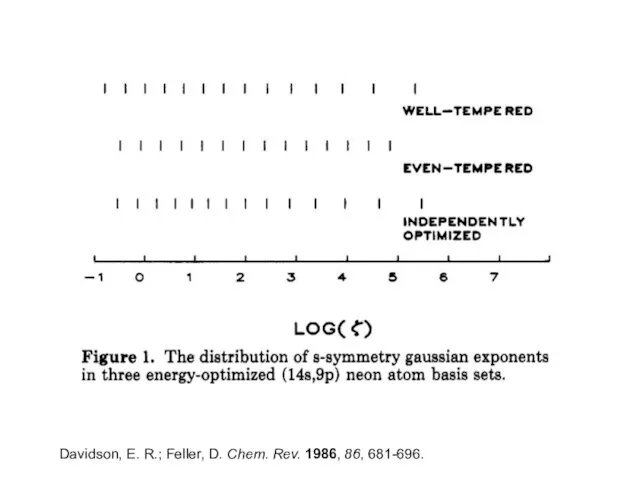
Davidson, E. R.; Feller, D. Chem. Rev. 1986, 86, 681-696.
Davidson, E. R.; Feller, D. Chem. Rev. 1986, 86, 681-696.

Used to model infinite systems (e.g. metals, crystals, etc.)
In infinite systems,
Used to model infinite systems (e.g. metals, crystals, etc.)
In infinite systems,

Polarization Functions
Similar exponent as valence function
Higher angular momentum (l+1)
Uncontracted Gaussian (coefficient=1)
Introduces
Polarization Functions
Similar exponent as valence function
Higher angular momentum (l+1)
Uncontracted Gaussian (coefficient=1)
Introduces

Diffuse Functions
Smaller exponent than valence functions
(larger spatial extent)
Same angular momentum
Diffuse Functions
Smaller exponent than valence functions
(larger spatial extent)
Same angular momentum

Cartesian vs. Spherical
Cartesians:
s – 1 function
p – 3 functions
d – 6
Cartesian vs. Spherical
Cartesians:
s – 1 function
p – 3 functions
d – 6

Cartesian vs. Spherical
Suppose we calculated the energy of HCl using a
Cartesian vs. Spherical
Suppose we calculated the energy of HCl using a

Pople Basis Sets
Optimized using Hartree-Fock
Names have the form
k-nlm++G** or k-nlmG(…)
k
Pople Basis Sets
Optimized using Hartree-Fock
Names have the form
k-nlm++G** or k-nlmG(…)
k

Pople Basis Sets
Examples:
6-31G Three contracted Gaussians for the core with the valence
Pople Basis Sets
Examples:
6-31G Three contracted Gaussians for the core with the valence

Dunning Correlatoin Consistent Basis Sets
Optimized using a correlated method (CIS, CISD,
Dunning Correlatoin Consistent Basis Sets
Optimized using a correlated method (CIS, CISD,

Dunning Basis Sets
Examples:
cc-pVDZ Double zeta with polarization
aug-cc-pVTZ Triple zeta with polarization and
diffuse
Dunning Basis Sets
Examples:
cc-pVDZ Double zeta with polarization
aug-cc-pVTZ Triple zeta with polarization and
diffuse

Extrapolate to complete basis set limit
Most useful for electron correlation methods
P(lmax)
Extrapolate to complete basis set limit
Most useful for electron correlation methods
P(lmax)

Basis Set Superposition Error
Occurs when a basis function centered at one
Basis Set Superposition Error
Occurs when a basis function centered at one

Counterpoise Correction
E(A)ab is the energy of fragment A with the basis
Counterpoise Correction
E(A)ab is the energy of fragment A with the basis

Additional Information
EMSL Basis Set Exchange:
https://bse.pnl.gov/bse/portal
Further reading:
Davidson, E. R.; Feller, D. Chem.
Additional Information
EMSL Basis Set Exchange:
https://bse.pnl.gov/bse/portal
Further reading:
Davidson, E. R.; Feller, D. Chem.

Effective Core Potentials (ECPs) and Model Core Potentials (MCPs)
Effective Core Potentials (ECPs) and Model Core Potentials (MCPs)

Frozen Core Approximation
Approximation made: atomic core orbitals are not allowed to
change
Frozen Core Approximation
Approximation made: atomic core orbitals are not allowed to
change

Pseudopotentials - ECPs
Effective core potentials (ECPs) are pseudopotentials that
replace core electrons
Pseudopotentials - ECPs
Effective core potentials (ECPs) are pseudopotentials that
replace core electrons

Shape Consistent ECPs
Nodeless pseudo-orbitals that resemble the valence orbitals in
Shape Consistent ECPs
Nodeless pseudo-orbitals that resemble the valence orbitals in

Energy Consistent ECPs
Approach that tries to reproduce the low-energy atomic
Energy Consistent ECPs
Approach that tries to reproduce the low-energy atomic
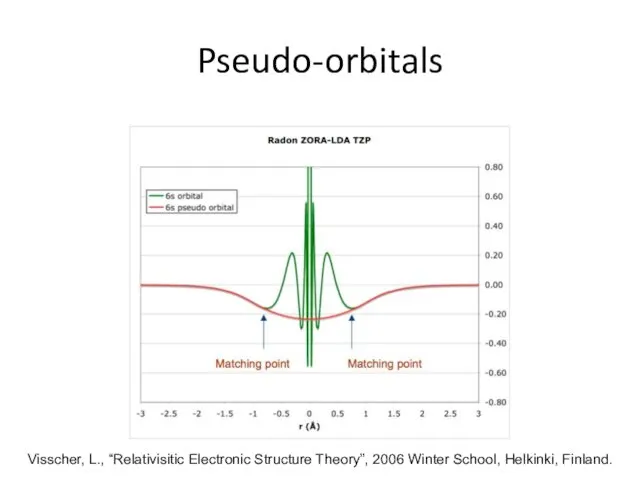
Pseudo-orbitals
Visscher, L., “Relativisitic Electronic Structure Theory”, 2006 Winter School, Helkinki, Finland.
Pseudo-orbitals
Visscher, L., “Relativisitic Electronic Structure Theory”, 2006 Winter School, Helkinki, Finland.
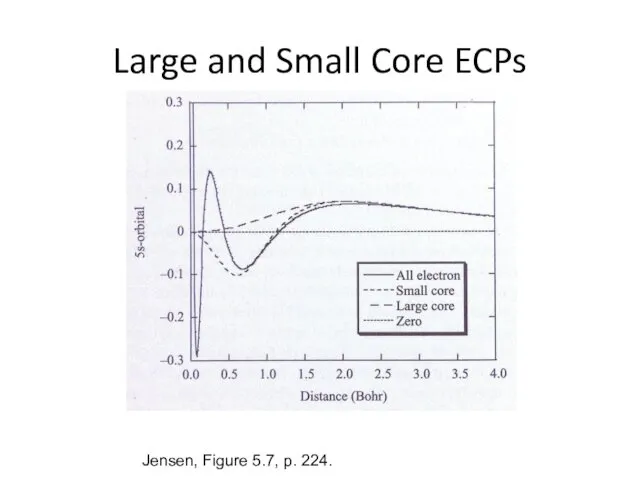
Large and Small Core ECPs
Jensen, Figure 5.7, p. 224.
Large and Small Core ECPs
Jensen, Figure 5.7, p. 224.

Pseudopotentials - MCPs
Model Core Potentials (MCP) provide a
computationally feasible
Pseudopotentials - MCPs
Model Core Potentials (MCP) provide a
computationally feasible

MCP Formulation
All-electron (AE) Hamiltonian:
MCP Hamiltonian:
First term is the 1 electron
MCP Formulation
All-electron (AE) Hamiltonian:
MCP Hamiltonian:
First term is the 1 electron

1-electron Hamiltonian
All-electron (AE) Hamiltonian:
MCP Hamiltonian:
First term is the 1 electron
1-electron Hamiltonian
All-electron (AE) Hamiltonian:
MCP Hamiltonian:
First term is the 1 electron

MCP Nuclear Attraction
AI, αI, BJ, and βJ are fitted MCP
MCP Nuclear Attraction
AI, αI, BJ, and βJ are fitted MCP
 Александр Евгеньевич Ферсман
Александр Евгеньевич Ферсман Кислород и озон
Кислород и озон Алкандар. Метан және оның құрылысы
Алкандар. Метан және оның құрылысы Соли аммония
Соли аммония Химические свойства алканов
Химические свойства алканов Свободное окисление и токсические формы кислорода
Свободное окисление и токсические формы кислорода Формирование ключевых и предметных компетенций учащихся при изучении темы “Металлы”
Формирование ключевых и предметных компетенций учащихся при изучении темы “Металлы” Углерод. Оксиды углерода
Углерод. Оксиды углерода Применение соляной кислоты и её солей
Применение соляной кислоты и её солей Антибиотики как ЛС
Антибиотики как ЛС Соли. CaSO4 - Сульфат кальция
Соли. CaSO4 - Сульфат кальция Class micro and macro elements
Class micro and macro elements Строение атома. Периодический закон Менделеева
Строение атома. Периодический закон Менделеева Простые вещества – неметаллы
Простые вещества – неметаллы Вычисление массовой доли растворенного вещества. 8 класс
Вычисление массовой доли растворенного вещества. 8 класс Цинк в функциональных пищевых и кормовых продуктах
Цинк в функциональных пищевых и кормовых продуктах Ювелирное дело. Империя самоцветов
Ювелирное дело. Империя самоцветов Ферменты в биотехнологии
Ферменты в биотехнологии Строение вещества и агрегатные состояния вещества
Строение вещества и агрегатные состояния вещества Мило та миловаріння
Мило та миловаріння Почему мыло пенится
Почему мыло пенится Основные классы неорганических соединений
Основные классы неорганических соединений Тренувальні вправи. Задачі
Тренувальні вправи. Задачі Період як особлива синтаксична конструкція
Період як особлива синтаксична конструкція Кислородсодержащие органические соединения. Лабораторная работа
Кислородсодержащие органические соединения. Лабораторная работа Химия элементов. Общая характеристика элементов
Химия элементов. Общая характеристика элементов Вклад М.В. Ломоносова в развитие химии
Вклад М.В. Ломоносова в развитие химии Узагальнення знань з теми Вуглеводні
Узагальнення знань з теми Вуглеводні