- Равновесие в растворах электролитов

Содержание
- 2. ЭЛЕКТРОХИМИЯ – это раздел физической химии, в котором изучается взаимосвязь между электрическими и химическими явлениями. Задачи
- 3. Теория Аррениуса - первая теория электролитической диссоциации Основные положения: Электролитами называются вещества, которые в растворе или
- 4. Сила электролита зависит: от природы растворителя от природы электролита Степень диссоциации зависит: от температуры присутствия других
- 5. Пример. Для 1 валентного электролита АВ с исходной концентрацией С равновесие при диссоциации имеет вид: АВ
- 6. С учетом понятия разведения, Закон Оствальда примет следующий вид: Разведение (V=1/С) – это объём раствора, содержащего
- 7. Концентрации ионов в растворе: Концентрации ионов водорода (Н+) и гидроксильной группы (ОН-) служат для характеристики среды
- 8. Связь степени диссоциации и изотонического коэффициента До создания теории Аррениуса, Вант - Гофф ввел понятие изотонического
- 9. Теория Аррениуса применима: только для разбавленных растворов слабых электролитов. Теория Аррениуса НЕ применима: для концентрированных растворов
- 10. Основной недостаток теории Аррениуса: пренебрежение электростатическим взаимодействием ионов в растворе, а также взаимодействием ионов с молекулами
- 11. Понятие активности вводится для оценки отклонения реальных растворов от идеальных. (1.11) Если в системе преобладают силы
- 12. Если молекула электролита диссоциирует на ν+ катионов и ν- анионов, то общая активность электролита α общ
- 13. Моляльная шкала концентраций в термодинамике растворов электролитов Средние величины коэффициента активности и моляльности представляют собой среднее
- 14. Для средней активности получаем расчетную формулу: (1.20) Выводы: Введение понятий о средней активности и среднем коэффициенте
- 15. Основные положения теории Дебая – Гюккеля: Раствор электролита представляет собой не механическую смесь катионов и анионов,
- 16. При математическом описании теории Дебай и Гюккель получили уравнение для эффективного радиуса ионной атмосферы (1/χ или
- 17. Примечание: Эффективный радиус ионной атмосферы зависит от концентрации электролита от температуры заряда ионов от природы растворителя
- 18. Количественные выражения среднего коэффициента активности зависят от концентрации растворов и носят название I, II и III
- 19. Из уравнения (1.22) видно, что средний коэффициент активности (γ±) зависит от ионной силы раствора, т.е. от
- 20. II приближение теории С увеличением концентрации растворов ионы приближаются друг к другу, и возникает необходимость учета
- 21. III приближение теории Для концентрированных растворов с ионной силой J ≤ 1 справедливо III приближение теории,
- 22. Зависимости lgγ± от ионной силы раствора в соответствии с I, II и III приближениями теории Дебая
- 23. Примечание: Если разрушаются сольватные оболочки ионов, то в растворе может возникнуть очень сильное отталкивание между ионами
- 24. В теории Кузнецовой энергия взаимодействия ионов описана с позиций ионного кристалла. Это позволило получить простые соотношения
- 25. Основное различие уравнений (1.26) и (1.28) в зависимостях логарифма среднего коэффициента активности от m1/2 и m1/3.
- 26. Расчетные формулы для электролитов различного типа (по теории Е. М. Кузнецовой ) Для электролита типа 1,1
- 28. Выводы (Из таблицы расчетных значений γ±): теория Е.М. Кузнецовой даёт хорошо согласующиеся с опытными данными результаты
- 29. Электрохимия изучает закономерности взаимных превращений электрической и химической энергии и процессы, связанные с прохождением электрического тока
- 31. Скачать презентацию

ЭЛЕКТРОХИМИЯ –
это раздел физической химии,
в котором изучается взаимосвязь между
ЭЛЕКТРОХИМИЯ –
это раздел физической химии,
в котором изучается взаимосвязь между

Теория Аррениуса - первая теория электролитической диссоциации
Основные положения:
Электролитами называются вещества,
Теория Аррениуса - первая теория электролитической диссоциации
Основные положения:
Электролитами называются вещества,

Сила электролита зависит:
от природы растворителя
от природы электролита
Сила электролита зависит:
от природы растворителя
от природы электролита

Пример. Для 1 валентного электролита АВ с исходной концентрацией С
равновесие
Пример. Для 1 валентного электролита АВ с исходной концентрацией С
равновесие

С учетом понятия разведения, Закон Оствальда примет следующий вид:
Разведение (V=1/С) –
С учетом понятия разведения, Закон Оствальда примет следующий вид:
Разведение (V=1/С) –

Концентрации ионов в растворе:
Концентрации ионов водорода (Н+) и гидроксильной группы (ОН-)
Концентрации ионов в растворе:
Концентрации ионов водорода (Н+) и гидроксильной группы (ОН-)

Связь степени диссоциации и изотонического коэффициента
До создания теории Аррениуса, Вант
Связь степени диссоциации и изотонического коэффициента
До создания теории Аррениуса, Вант

Теория Аррениуса применима:
только для разбавленных растворов слабых электролитов.
Теория Аррениуса
Теория Аррениуса применима:
только для разбавленных растворов слабых электролитов.
Теория Аррениуса

Основной недостаток теории Аррениуса:
пренебрежение электростатическим взаимодействием ионов в растворе,
Основной недостаток теории Аррениуса:
пренебрежение электростатическим взаимодействием ионов в растворе,

Понятие активности вводится для оценки отклонения реальных растворов от идеальных.
(1.11)
Если
Понятие активности вводится для оценки отклонения реальных растворов от идеальных.
(1.11)
Если

Если молекула электролита диссоциирует на ν+ катионов и ν- анионов, то
Если молекула электролита диссоциирует на ν+ катионов и ν- анионов, то

Моляльная шкала концентраций в термодинамике растворов электролитов
Средние величины коэффициента активности
Моляльная шкала концентраций в термодинамике растворов электролитов
Средние величины коэффициента активности

Для средней активности получаем расчетную формулу:
(1.20)
Выводы:
Введение понятий о средней
Для средней активности получаем расчетную формулу:
(1.20)
Выводы:
Введение понятий о средней

Основные положения теории Дебая – Гюккеля:
Раствор электролита представляет собой не
Основные положения теории Дебая – Гюккеля:
Раствор электролита представляет собой не

При математическом описании
теории Дебай и Гюккель получили
уравнение для эффективного
При математическом описании
теории Дебай и Гюккель получили
уравнение для эффективного

Примечание:
Эффективный радиус ионной атмосферы зависит
от концентрации электролита
от температуры
заряда ионов
от
Примечание:
Эффективный радиус ионной атмосферы зависит
от концентрации электролита
от температуры
заряда ионов
от

Количественные выражения среднего коэффициента активности
зависят от концентрации растворов и носят
Количественные выражения среднего коэффициента активности
зависят от концентрации растворов и носят

Из уравнения (1.22) видно, что средний коэффициент активности (γ±)
зависит от
Из уравнения (1.22) видно, что средний коэффициент активности (γ±)
зависит от

II приближение теории
С увеличением концентрации растворов ионы приближаются друг к другу,
II приближение теории
С увеличением концентрации растворов ионы приближаются друг к другу,

III приближение теории
Для концентрированных растворов с ионной силой J ≤ 1
III приближение теории
Для концентрированных растворов с ионной силой J ≤ 1
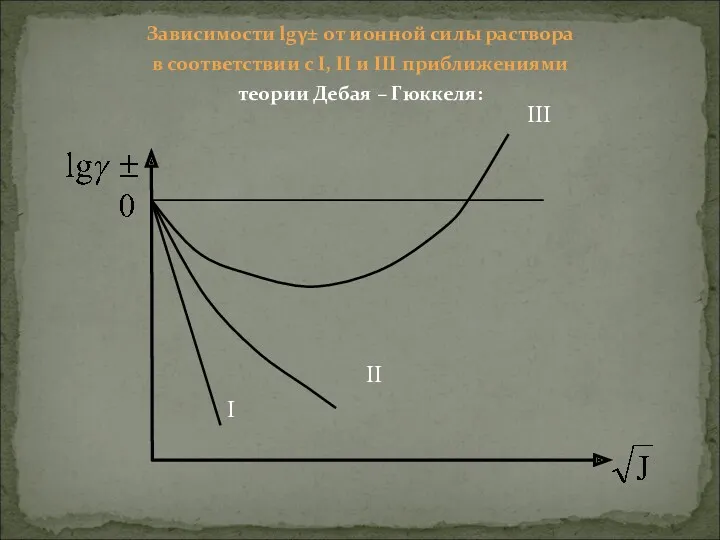
Зависимости lgγ± от ионной силы раствора
в соответствии с I, II
Зависимости lgγ± от ионной силы раствора
в соответствии с I, II

Примечание:
Если разрушаются сольватные оболочки ионов, то в растворе может возникнуть очень
Примечание:
Если разрушаются сольватные оболочки ионов, то в растворе может возникнуть очень

В теории Кузнецовой энергия взаимодействия ионов описана с позиций ионного кристалла.
В теории Кузнецовой энергия взаимодействия ионов описана с позиций ионного кристалла.

Основное различие уравнений (1.26) и (1.28) в зависимостях логарифма среднего коэффициента
Основное различие уравнений (1.26) и (1.28) в зависимостях логарифма среднего коэффициента

Расчетные формулы для электролитов различного типа (по теории Е. М.
Расчетные формулы для электролитов различного типа (по теории Е. М.
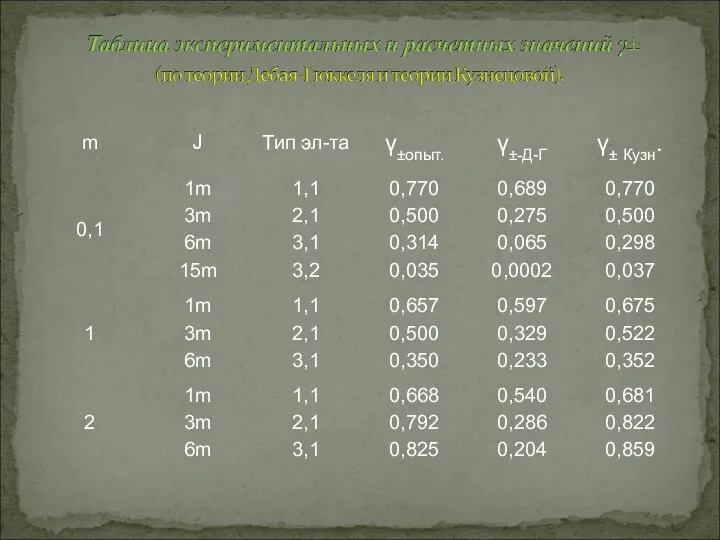

Выводы (Из таблицы расчетных значений γ±):
теория Е.М. Кузнецовой даёт хорошо согласующиеся
Выводы (Из таблицы расчетных значений γ±):
теория Е.М. Кузнецовой даёт хорошо согласующиеся

Электрохимия изучает закономерности взаимных превращений электрической и химической энергии и процессы,
Электрохимия изучает закономерности взаимных превращений электрической и химической энергии и процессы,
 Одноатомные спирты
Одноатомные спирты Обмен липидов
Обмен липидов Органическая химия. Подготовка к контрольной работе № 2
Органическая химия. Подготовка к контрольной работе № 2 Чистые вещества и смеси
Чистые вещества и смеси Водород. Химический элемент
Водород. Химический элемент Полимеры. Пластмассы. Волокна
Полимеры. Пластмассы. Волокна λ-MnO2 as material with pseudocapacitive properties
λ-MnO2 as material with pseudocapacitive properties Повышение эффективности производства изопропилбензола за счёт нового катализатора, производительность по кумолу 100500 т/год
Повышение эффективности производства изопропилбензола за счёт нового катализатора, производительность по кумолу 100500 т/год Углеводороды. Значение углеводородов
Углеводороды. Значение углеводородов Мембранный транспорт ионов: электродиффузионная теория
Мембранный транспорт ионов: электродиффузионная теория Полімери, Їх властивості та застосування
Полімери, Їх властивості та застосування Оксид фосфора
Оксид фосфора Углерод. Аллотропные состояния углерода
Углерод. Аллотропные состояния углерода Железо и его сплавы
Железо и его сплавы Химическая посуда и лабораторное оборудование
Химическая посуда и лабораторное оборудование Гигиена питания школьников
Гигиена питания школьников Синтетические топлива
Синтетические топлива Electrolysis
Electrolysis Строение и свойства циклоалканов
Строение и свойства циклоалканов Катионы І - ІІІ аналитических групп
Катионы І - ІІІ аналитических групп Створення 3D моделей атомів й молекул елементів
Створення 3D моделей атомів й молекул елементів Периодическая система химических элементов. Периоды
Периодическая система химических элементов. Периоды Лекция 3. Гидроксисоединения. Карбонильные соединения
Лекция 3. Гидроксисоединения. Карбонильные соединения Prezentatsia
Prezentatsia Диссоциация кислот, оснований, солей
Диссоциация кислот, оснований, солей Кислород и его свойства
Кислород и его свойства Электролиз. 11 класс
Электролиз. 11 класс Растворы. Процесс растворения
Растворы. Процесс растворения